
QIIME 2 2021.2 版本现已推…
QIIME 2 2021.2 版本现已推…
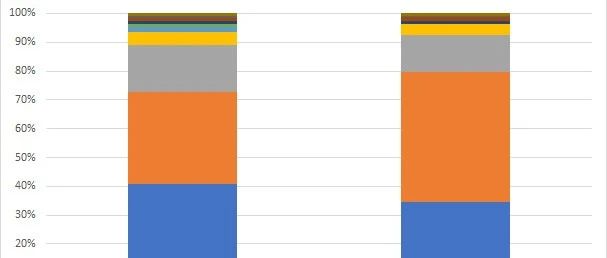
最早接触Kraken2这个软件是在宏基因…

前面已经完成了qiime2-sidle插…

前面说到Science封面文章用的16S…

继续前面的文档学习,地址在这里啦!官方文…

前面介绍的SMURF流程的运行以失败告终…

肠道微生物是近两年的研究热点,但是去年登…

3.4 微生物数据组成分析 早在1897…
最近做PRS评分,需要用到连锁不平衡信息,来进行位点筛选,找了好几个工具用于计算连锁不平衡,也发现了个好用的网页工具来进行查询,在这里和大家分享一下!
地址在这,阅读原文也是这个,后面的操作都在这个网页进行的。由于这个网站用到了谷歌的一些组件,可能需要科学上网才能访问。什么,你不会,我有个凑活着用的方式,要不要试试,回复”setup”,给你个小工具。
发现这个软件之前的官网已经打不开,但是在github上仍然在更新,https://github.com/SyntekabioTools/HLAscan或许是换了工作?最近一次更新是2019.12.4,还是比较新的。发现wegene的NGS HLA分型报告是用的这个软件的参考文献,估计还是权威些的。