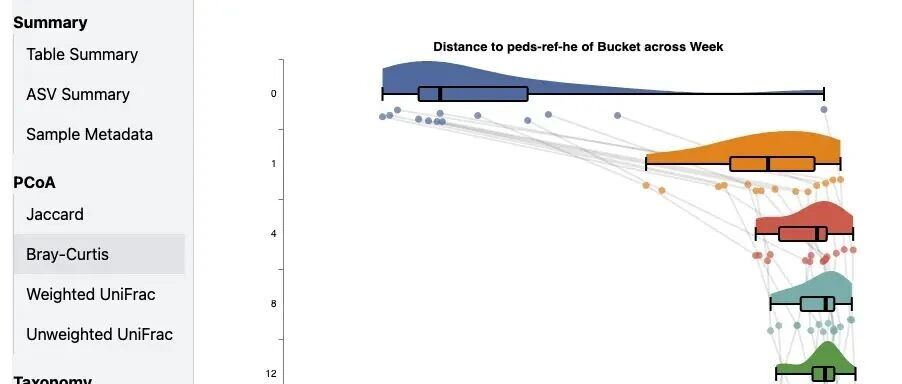
大家好!QIIME 2 2…
大家好!QIIME 2 2…

小编荐语:这个数据库厉害了…
最大的公开人线粒体变异数据库HelixM…

最近想要查找微生物中文名的时候发现一个宝…
AI能力的发展一日千里呀,…

小编导读 微生物研究领域一…
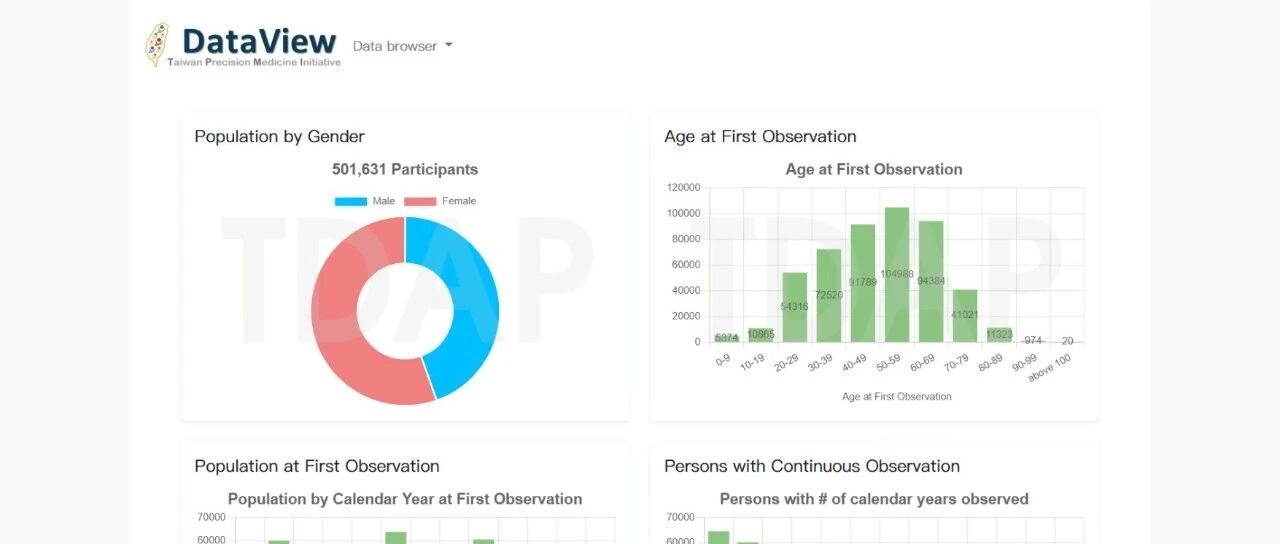
小编发现台湾精准医疗计划(TPMI)的成…
QIIME 2 2025.7 QIIME…
通过分析 59 种疾病和健康队列的蛋白质…

大家好!今天我们来分享一篇发表在《Com…