
R、RStudio和ggplot2简介 …
R、RStudio和ggplot2简介 …

4.3 ggplot2简介 4.3.1 …

最近读微生态公众号中宏基因组的文章,发现…

一、 R启动文件 每次R语言启动读入.R…

这是《高效R语言编程》的学习笔记,前面的…

这是《高效R语言编程》的学习笔记,前面的…

小编提示: 最值得关注的几点: 1、qi…
最近从南图借了本《高效R语言编程》,记些…

发现如果习惯了一个编程语言,想当然的往另…
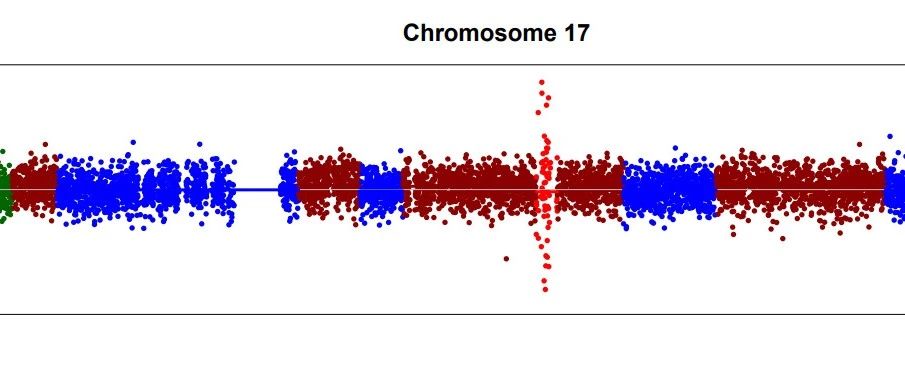
试用了两个软件用于测试CNV检测,虽然没…