群体遗传学领域,看起来华人(中国人)至少是占据前线的,至少很多软件的一作就是中国人的名字,至少活主要是我们干的。当然,也有很多华人(国人)大牛开发了史诗级别的软件,向勤劳的华夏人致敬!SAIGE 这个软件也是如此。最新更新了1.0版,一起来学习下新的软件文档。
总览
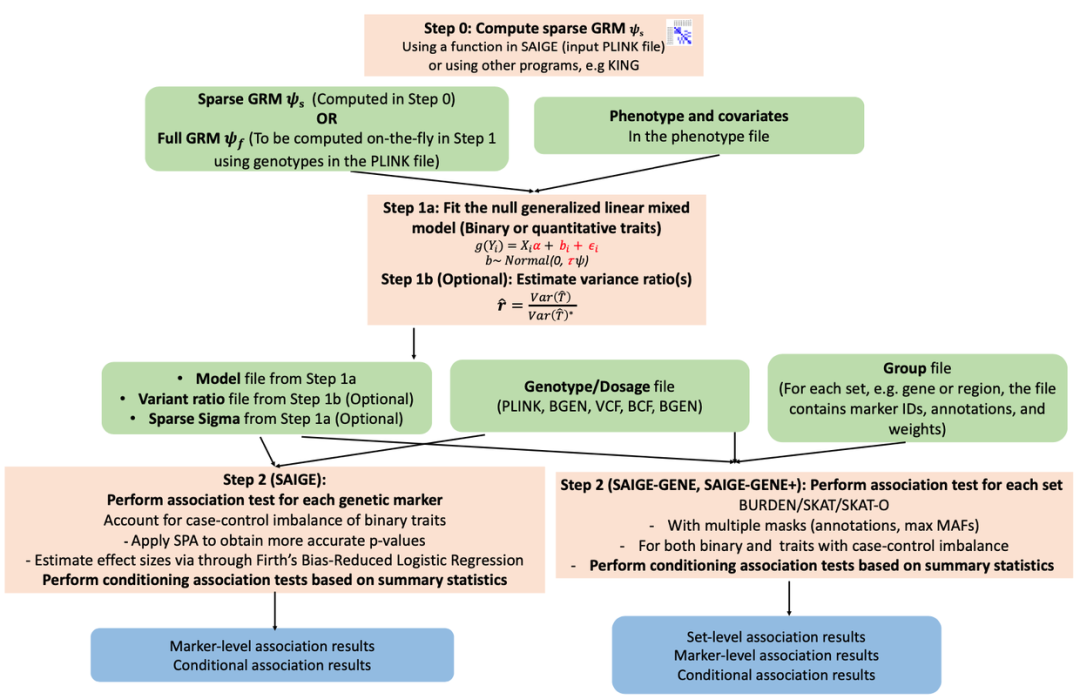
简介
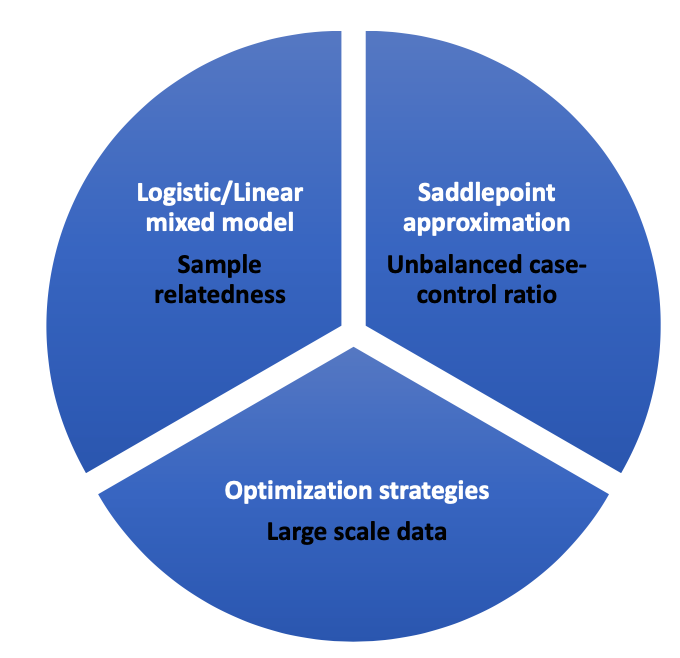
SAIGE是与Rcpp一起开发的R包,用于大规模数据集和生物库中的全基因组关联测试。方法
-
基于广义混合模型的样本相关性 -
允许使用全遗传关系矩阵或稀疏遗传关系矩阵(GRM)进行模型拟合 -
适用于数量和二元分类性状 -
处理二元性状的病例-对照不平衡 -
对于大型数据集,计算效率高 -
执行单变异关联测试 -
通过 Firth 偏倚减少逻辑回归提供效应大小估计 -
执行条件关联分析
SAIGE-GENE(现在称为SAIGE-GENE+)是R包中的新方法扩展,用于基于集合的罕见变异分析。
-
执行 BURDEN、SKAT 和 SKAT-O 分析 -
允许对多个次要等位基因频率阀值和功能注释进行分析 -
允许在基于集合的分析中指定标记的权重 -
执行条件分析以识别独立于近 GWAS 信号的关联
该软件包采用以下格式的基因型文件输入
-
PLINK (bed, bim, fam), BGEN, VCF, BCF, SAV
引用
SAIGE:
-
Zhou W, Nielsen JB, Fritsche LG, Dey R, Gabrielsen ME, Wolford BN, LeFaive J, VandeHaar P, Gagliano SA, Gifford A, Bastarache LA, Wei WQ, Denny JC, Lin M, Hveem K, Kang HM, Abecasis GR, Willer CJ, Lee S. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet. 2018 Sep;50(9):1335-1341. doi: 10.1038/s41588-018-0184-y. Epub 2018 Aug 13. PMID: 30104761; PMCID: PMC6119127.
SAIGE-GENE:
-
Zhou W*, Zhao Z*, Nielsen JB, Fritsche LG, LeFaive J, Gagliano Taliun SA, Bi W, Gabrielsen ME, Daly MJ, Neale BM, Hveem K, Abecasis GR, Willer CJ, Lee S. Scalable generalized linear mixed model for region-based association tests in large biobanks and cohorts. Nat Genet. 2020 Jun;52(6):634-639. doi: 10.1038/s41588-020-0621-6. Epub 2020 May 18. PMID: 32424355; PMCID: PMC7871731.
SAIGE-GENE+:
-
Wei Zhou*, Wenjian Bi*, Zhangchen Zhao*, Kushal K. Dey, Karthik A. Jagadeesh, Konrad J. Karczewski, Mark J. Daly, Benjamin M. Neale, Seunggeun Lee. Set-based rare variant association tests for biobank scale sequencing data sets. medRxiv 2021.07.12.21260400; doi: https://doi.org/10.1101/2021.07.12.21260400
许可证
SAIGE在GPL许可证下分发。
联系
如果您对SAIGE有任何疑问,请联系 saige.genetics@gmail.com
安装
如何安装和运行 SAIGE 和 SAIGE-GENE
安装 SAIGE/SAIGE-GENE(当前版本 1.0.0(更新于 2022 年 3 月 15 日))
依赖项列表:
-
R >= 3.6.1, gcc >= 5.4.0, cmake 3.14.1, cget[1], savvy[2]
-
R 包: Rcpp (>= 1.0.7), RcppArmadillo, RcppParallel, data.table, SPAtest (== 3.1.2), RcppEigen, Matrix, methods, BH, optparse, SKAT, qlcMatrix, RhpcBLASctl, roxygen2, rversions, devtools, dplyr, dbplyr
-
Rscript ./extdata/install_packages.R 可用于安装 R 包
-
两个用于读取 VCF 文件的库。将在 SAIGE 安装过程中自动安装
-
cget[3],savvy[4]
日志:
v1.0.1(2022 年 3 月 18 日):
修复错误: –AlleleOrder bgen 输入单变异关联分析输出的标题
改进:在 CCT 中将 PI 更改为M_PI.cpp在 Makevar 中添加标志以安装在 Centos 8 上
v1.0.0(2022 年 3 月 15 日):第一个稳定版本
v0.99.3(2022 年 3 月 12 日):对于基于集合的分析,所有与 –condition 一起使用的标记 ID 和组文件中都需要使用 chr:pos:ref:alt 格式
使用源代码 https://github.com/saigegit/SAIGE 安装 SAIGE
安装依赖项
R >= 3.6.1, gcc >= 5.4.0, cmake 3.14.1,从 github 下载 SAIGE
repo_src_url=https://github.com/saigegit/SAIGE
git clone --depth 1 -b $src_branch $repo_src_url
安装依赖项:R 包
Rscript ./SAIGE/extdata/install_packages.R
安装 SAIGE R 软件包
将 SAIGE 安装到存储所有 R 库的根目录
–library=path_to_final_SAIGE_library 可用于指定安装 SAIGE 的目录
R CMD INSTALL --library=path_to_final_SAIGE_library SAIGE
运行 SAIGE
如果未在根 R 库路径中安装 SAIGE,请更改 library(SAIGE)自
library(SAIGE, lib.loc="path_to_final_SAIGE_library")在以下脚本中
SAIGE/extdata/step1_fitNULLGLMM.R
SAIGE/extdata/step2_SPAtests.R
SAIGE/extdata/createSparseGRM.R
使用 conda 环境安装 SAIGE
-
下载并安装[miniconda[5]](https://docs.conda.io/en/latest/miniconda.html)
-
使用
conda env create -f environment-RSAIGE.yml -
conda 环境文件位于 SAIGE 文件夹中:./conda_env/environment-RSAIGE.yml
-
下载环境-RSAIGE.yml 后,运行以下命令
-
激活 CONDA 环境 RSAIGE
conda activate RSAIGE
FLAGPATH=`which python | sed 's|/bin/python$||'`
export LDFLAGS="-L${FLAGPATH}/lib"
export CPPFLAGS="-I${FLAGPATH}/include"请确保使用导出(最后两个命令行)设置LDFLAGS和CPPFLAGS,以便在编译SAIGE源代码时可以正确链接库。
注意:以下是[6]创建 conda 环境文件的步骤
-
从源代码安装 SAIGE。
src_branch=main
repo_src_url=https://github.com/saigegit/SAIGE
git clone --depth 1 -b $src_branch $repo_src_url
Rscript ./SAIGE/extdata/install_packages.R
R CMD INSTALL --library=path_to_final_SAIGE_library SAIGE当在 R 中调用 SAIGE 时,设置 lib.loc=path_to_final_SAIGE_library
library(SAIGE, lib.loc=path_to_final_SAIGE_library)
使用 Docker 映像运行 SAIGE
感谢 Juha Karjalainen 分享 Dockerfile。
Dockerfile 可以在 SAIGE 文件夹中找到:./docker/Dockerfile
可以拉取映像docker pull wzhou88/saige:1.0.0可以调用函数
docker run --rm -it -v /data/haod/:/home wzhou88/saige:1.0.0
cd /home
step1_fitNULLGLMM.R --help
step2_SPAtests.R --help
createSparseGRM.R --help
### 从 bioconda 安装 SAIGE (不是当前版本)
警告:请不要将此 BIOCONDA 版本用于 BGEN 输入。我们正在研究这个问题。

要从 conda 安装 saige,只需使用最新版本的 R 和 saige 创建环境:
conda create -n saige -c conda-forge -c bioconda "r-base>=4.0" r-saige
conda activate saige
有关 r-saige conda 软件包[7]和可用版本的更多信息,请参阅问题 #272[8]。
单变异关联分析
SAIGE采取两个步骤来执行单变异关联分析
-
我们建议对 MAC >= 20 的变体进行单变体关联分析 -
对于**罕见的变异关联,请使用 SAIGE-GENE+**进行基于集合的关联分析
步骤 1:拟合null(空)逻辑/线性混合模型
-
对于二元性状,将拟合一个空逻辑混合模型(–traitType=binary)。
-
对于定量性状,将拟合空线性混合模型(*-traitType=quantitative),并且需要逆归一化(–invNormalize=TRUE*)
-
默认情况下,协变量作为补偿包含在模型中,请使用
-isCovariateOffset=FALSE停用此功能。这将花费更多的计算时间与-isCovariateOffset=FALSE -
示例脚本位于 extdata 文件夹中
#go to the folder
cd extdata
#check the help info for step 1
Rscript step1_fitNULLGLMM.R --help
使用完整 GRM 拟合空模型
对于二元性状,将拟合一个空逻辑混合模型(–TRAITTYPE=BINARY)。
-
使用 -qCovarColList 列出分类协变量。将为不同的级别创建虚拟变量(独热编码)。 -
使用 -nThreads表示要使用的 CPU 数量
Rscript step1_fitNULLGLMM.R
--plinkFile=./input/nfam_100_nindep_0_step1_includeMoreRareVariants_poly_22chr
--phenoFile=./input/pheno_1000samples.txt_withdosages_withBothTraitTypes.txt
--phenoCol=y_binary
--covarColList=x1,x2,a9,a10
--qCovarColList=a9,a10
--sampleIDColinphenoFile=IID
--traitType=binary
--outputPrefix=./output/example_binary
--nThreads=24
--IsOverwriteVarianceRatioFile=TRUE
对于定量性状,将拟合空线性混合模型(–TRAITTYPE=定量),并且需要逆归一化(–INVNORMALIZE=TRUE)
Rscript step1_fitNULLGLMM.R
--plinkFile=./input/nfam_100_nindep_0_step1_includeMoreRareVariants_poly_22chr
--useSparseGRMtoFitNULL=FALSE
--phenoFile=./input/pheno_1000samples.txt_withdosages_withBothTraitTypes.txt
--phenoCol=y_quantitative
--covarColList=x1,x2,a9,a10
--qCovarColList=a9,a10
--sampleIDColinphenoFile=IID
--invNormalize=TRUE
--traitType=quantitative
--outputPrefix=./output/example_quantitative
--nThreads=24
--IsOverwriteVarianceRatioFile=TRUE
使用稀疏 GRM 拟合空模型(将仅使用一个 CPU)
参考GWASLab的文章,这个方法不推荐,因为没有LOCO。
-
Use –useSparseGRMtoFitNULL=TRUE
-
对包含稀疏 GRM 的文件使用 –稀疏 GRMFile
-
对包含稀疏 GRM 中样本 ID 的文件使用 –稀疏 GRM 样本 ID
-
如果未使用 –plinkFile 指定 plink 文件,则不会估计方差比,请确保在步骤 2 中指定 –sparseSigmaFile(由步骤 1 输出)以进行关联测试
-
要估计方差比并在步骤 2 中使用,请指定包含将用于使用 –plinkFile 进行方差比估计的标记的 plink 文件。例如 –plinkFile=./input/nfam_100_nindep_0_step1_includeMoreRareVariants_poly_22chr 和 –skipVarianceRatioEstimation=FALSE 注意:这将加快步骤 2 的速度
-
要仅包含样本子集以拟合空模型,请使用 –SampleIDIncludeFile
-
将仅使用一个 CPU,并且不会应用 LOCO
Rscript step1_fitNULLGLMM.R
--sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx
--sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt
--useSparseGRMtoFitNULL=TRUE
--phenoFile=./input/pheno_1000samples.txt_withdosages_withBothTraitTypes.txt
--phenoCol=y_binary
--covarColList=x1,x2,a9,a10
--qCovarColList=a9,a10
--sampleIDColinphenoFile=IID
--traitType=binary
--outputPrefix=./output/example_binary_sparseGRM
输入文件
-
(必填)表型文件(包含协变量(如果有),如性别和年龄)文件可以是空格,也可以是用标题以制表符分隔的。该文件必须包含一列用于样本 ID,一列用于表型。它可能包含协变量列。
less -S ./input/pheno_1000samples.txt_withdosages_withBothTraitTypes.txt

-
(可选)用于构建完整遗传关系矩阵(GRM)和估计方差比的基因型 文件 SAIGE 采用基因型的 PLINK 二进制文件,并假设文件前缀与 .bed, .bim和 .fam的文件前缀相同。
./input/nfam_100_nindep_0_step1_includeMoreRareVariants_poly.bed
./input/nfam_100_nindep_0_step1_includeMoreRareVariants_poly.bim
./input/nfam_100_nindep_0_step1_includeMoreRareVariants_poly.fam
-
(可选)稀疏 GRM 文件和稀疏 GRM 的示例 ID 文件。有关更多详细信息,请参阅步骤 0。
./output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx
./output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt
##the sparse matrix can be read and viewed using the R library Matrix
library(Matrix)
sparseGRM = readMM("./output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx")
输出文件
-
模型文件(步骤 2 的输入))
./output/example_quantitative.rda
#load the model file in R
load("./output/example_quantitative.rda")
names(modglmm)
modglmm$theta
输出 -
方差比文件(如果在步骤 1 中估计了方差比,则将在步骤 2 中输入)
less -S ./output/example_quantitative.varianceRatio.txt
1.21860521240473 -
随机选择的标记子集的关联结果文件(如果在步骤 1 中估计了方差比,则将生成此文件。它是一个中间文件,后续步骤不需要它。)
less -S ./output/example_quantitative_30markers.SAIGE.results.txt
markerIndex p.value p.value.NA var1 var2 Tv1 N AC AF
69970 0.214147816412411 0.173552665987772 1.14375062050603 0.953027027027038
1.32853003974658 1000 74 0.037
96109 0.953143629473153 0.949713298740539 1.07324590784922 0.931653266331636
-0.0608734702523558 1000 199 0.0995
步骤 2:执行单变体关联测试
-
对于二元性状,鞍点近似用于解释病例–对照不平衡。 -
可以使用要分析的遗传变异的剂量/基因型的文件格式:PLINK,VCF,BGEN[9],SAV[10] -
可以在步骤 2 中执行基于条件分析的汇总统计信息(–condition) -
查询和测试标记子集 -
-
变体 ID (chr:pos_ref/alt) 和染色体位置范围(chr 开始 结束)都可以为 BGEN 输入指定 (-idstoIncludeFile, -rangestoIncludeFile) -
-markers_per_chunk可用于指定要测试的标记数并将其输出为一个块。默认值 = 10000。请注意,小数字可能会减慢运行速度。要求此数字为 > = 1000。 -
如果 LOCO=TRUE(默认情况下),则必须指定–chrom,因此只有基因型/剂量文件应仅包含一条染色体 -
对于 VCF/BCF/SAV 输入,-vcfField=DS 用于剂量,-vcfField=GT 用于分析基因型 -
默认情况下,缺失的基因型/剂量将被归因为最佳猜测的 gentoypes/剂量(如 round(2xfreq),–impute_method=best_guess)。请注意,目前不支持删除缺少基因型/剂量的样本 -
–sampleFile 用于为 bgen 文件指定具有示例 ID 的文件。请不要在文件中包含表头。
#check the help info for step 2
Rscript step2_SPAtests.R --help
-
对于二元性状,使用 –is_output_moreDetails=TRUE 输出病例和对照组中的杂合子和纯合子计数
如果在步骤 1 中估计了方差比 (–varianceRatioFile=)
-
Using –bgenFile, –bgenFileIndex, –AlleleOrder, –sampleFile for bgen input
Rscript step2_SPAtests.R
--bgenFile=./input/genotype_100markers.bgen
--bgenFileIndex=./input/genotype_100markers.bgen.bgi
--sampleFile=./input/samplelist.txt
--AlleleOrder=ref-first
--SAIGEOutputFile=./output/genotype_100markers_marker_bgen_Firth.txt
--chrom=1
--minMAF=0
--minMAC=20
--GMMATmodelFile=./output/example_binary.rda
--varianceRatioFile=./output/example_binary.varianceRatio.txt
--is_Firth_beta=TRUE
--pCutoffforFirth=0.1
-
使用 –bedFile, –bimFile, –famFile, –AlleleOrder进行 PLINK 输入
Rscript step2_SPAtests.R
--bedFile=./input/genotype_100markers.bed
--bimFile=./input/genotype_100markers.bim
--famFile=./input/genotype_100markers.fam
--AlleleOrder=alt-first
--SAIGEOutputFile=./output/genotype_100markers_marker_plink.txt
--chrom=1
--minMAF=0
--minMAC=20
--GMMATmodelFile=./output/example_binary.rda
--varianceRatioFile=./output/example_binary.varianceRatio.txt
--is_output_moreDetails=TRUE
-
使用 –vcfFile、 –vcfFileIndex、 –vcfField 进行 VCF、BCF 和 SAV 输入
Rscript step2_SPAtests.R
--vcfFile=./input/genotype_100markers.vcf.gz
--vcfFileIndex=./input/genotype_100markers.vcf.gz.csi
--vcfField=GT
--SAIGEOutputFile=./output/genotype_100markers_marker_vcf.txt
--chrom=1
--minMAF=0
--minMAC=20
--GMMATmodelFile=./output/example_binary.rda
--varianceRatioFile=./output/example_binary.varianceRatio.txt
--is_output_moreDetails=TRUE
如果在步骤 1 中未估计方差比,因为使用了稀疏 GRM 来拟合空模型(与步骤 1 中相同,–sparseGRMFile and –sparseGRMSampleIDFile 用于指定稀疏 GRM 和样本 ID)
Rscript step2_SPAtests.R
--vcfFile=./input/genotype_100markers.vcf.gz
--vcfFileIndex=./input/genotype_100markers.vcf.gz.csi
--vcfField=GT
--SAIGEOutputFile=./output/genotype_100markers_marker_vcf_step1withSparseGRM.txt
--chrom=1
--minMAF=0
--minMAC=20
--GMMATmodelFile=./output/example_binary_sparseGRM.rda
--is_output_moreDetails=TRUE
--sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx
--sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt
对于二元性状,可以通过Firth的偏差减少逻辑回归更准确地估计效应大小,方法是设置
–is_Firth_beta=TRUE 和 –pCutoffforFirth=0.01
-
p 值为 <= pCutoffforFirth 的标记的效应大小将通过Firth 的偏倚减少逻辑回归进行估计
Rscript step2_SPAtests.R
--vcfFile=./input/genotype_100markers.vcf.gz
--vcfFileIndex=./input/genotype_100markers.vcf.gz.csi
--vcfField=GT
--SAIGEOutputFile=./output/genotype_100markers_marker_vcf_step1withSparseGRM.txt
--chrom=1
--minMAF=0
--minMAC=20
--GMMATmodelFile=./output/example_binary_sparseGRM.rda
--is_output_moreDetails=TRUE
--sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx
--sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt
--is_Firth_beta=TRUE
--pCutoffforFirth=0.01
条件分析
-
-condition = 冒号分隔的变异id (chr:pos:ref:alt) 比如:chr3:101651171:C:T,chr3:101651186:G:A
Rscript step2_SPAtests.R
--vcfFile=./input/genotype_100markers.vcf.gz
--vcfFileIndex=./input/genotype_100markers.vcf.gz.csi
--vcfField=GT
--SAIGEOutputFile=./output/genotype_100markers_marker_vcf_cond.txt
--chrom=1
--minMAF=0
--minMAC=20
--GMMATmodelFile=./output/example_binary.rda
--varianceRatioFile=./output/example_binary.varianceRatio.txt
--is_output_moreDetails=TRUE
--condition=1:13:A:C,1:79:A:C
输入文件
-
(必需)剂量文件 SAIGE 支持不同的剂量格式: PLINK, VCF, BCF, BGEN[11] 和 SAV[12].
./input/genotype_100markers.bed
./input/genotype_100markers.bim
./input/genotype_100markers.fam./input/genotype_100markers.bgen
./input/genotype_100markers.bgen.bgi#go to input
cd ./input
#index file can be generated using tabix
tabix --csi -p vcf genotype_10markers.vcf.gz
zcat genotype_10markers.vcf.gz | less -S
genotype_10markers.vcf.gz.csi#index file can be generated using tabix
tabix --csi -p vcf dosage_10markers.vcf.gz
zcat dosage_10markers.vcf.gz | less -S
dosage_10markers.vcf.gz.csidosage_10markers.sav
dosage_10markers.sav.s1r -
SAV -
含剂量的vcf -
含有基因型的VCF -
BGEN -
plink -
(可选。仅适用于不包含样品 ID 的 BGEN 文件) 示例文件包含一列用于与剂量文件中的示例顺序相对应的示例 ID。不包括标头。该选项最初用于不包含示例信息的 BGEN 文件。
less -S ./input/samplelist.txt
1a1
1a2
1a3 -
(必需。步骤 1) 步骤 1 中的模型文件输出
./output/example_binary.rda -
(可选。步骤 1) 步骤 1 中的方差比文件中的输出
./output/example_binary.varianceRatio.txt -
(可选。与步骤 1) 稀疏 GRM 文件中的输入和稀疏 GRM 的示例 ID 文件相同。有关更多详细信息,请参阅步骤 0。
./output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx
./output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt
##the sparse matrix can be read and viewed using the R library Matrix
library(Matrix)
sparseGRM = readMM("./output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx")
输出文件
-
包含关联分析结果的文件
less -S ./output/genotype_100markers_marker_vcf.txt
CHR POS MarkerID Allele1 Allele2 AC_Allele2 AF_Allele2 MissingRate
BETA SE Tstat var p.value p.value.NA Is.SPA p.value.NA_c Is.SPA.converge AF_case AF_ctrl N_case N_ctrl
1 4 rs4 A C 20 0.01 0 -0.145553 0.774596 -0.242588 1.66667 0.850949 0.850949 false 0.0102041 0.00997783 98 902 0 2 0 18 -
输出文件的索引。此文件包含关联测试的进度。如果在步骤 2 中指定 –is_overwrite_output=FALSE,程序将检查此索引并继续未完成的分析,而不是从头开始
less -S ./output/genotype_100markers_marker_vcf.txt.index
This is the output index file for SAIGE package to record the end point in case users want to restart the analysis. Please do not modify this file.
This is a Marker level analysis.
nEachChunk = 10000
Have completed the analysis of chunk 1
Have completed the analyses of all chunks.
注意:
-
关联结果与等位基因2有关。
#check the header
head -n 1 output/genotype_100markers_marker_vcf_cond.txt

CHR: chromosome
POS: genome position
SNPID: variant ID
Allele1: allele 1
Allele2: allele 2
AC_Allele2: allele count of allele 2
AF_Allele2: allele frequency of allele 2
MissingRate: missing rate (If the markers in the dosage/genotype input are imputed with is_imputed_data=TRUE, imputationInfo will be output instead of MissingRate)
imputationInfo: imputation info (output with is_imputed_data=TRUE). If not in dosage/genotype input file, will output 1
BETA: effect size of allele 2
SE: standard error of BETA
Tstat: score statistic of allele 2
var: estimated variance of score statistic
p.value: p value (with SPA applied for binary traits)
p.value.NA: p value when SPA is not applied (only for binary traits)
Is.SPA.converge: whether SPA is converged or not (only for binary traits)
#if --condition= is used for conditioning analysis, the conditional analysis results will be output
BETA_c, SE_c, Tstat_c, var_c, p.value_c, p.value.NA_c
AF_case: allele frequency of allele 2 in cases (only for binary traits)
AF_ctrl: allele frequency of allele 2 in controls (only for binary traits)
N_case: sample size in cases (only for binary traits)
N_ctrl: sample size in controls (only for binary traits)
if --is_output_moreDetails=TRUE
homN_Allele2_cases, hetN_Allele2_cases, homN_Allele2_ctrls, hetN_Allele2_ctrls will be output for sample sizes with different genotypes in cases and controls (only for binary traits)
示例 1
-
二元性状(–性状类型=二元) -
使用完整的 GRM 拟合空模型,该 GRM 将使用 plink 文件中的基因型动态计算 (–plinkFile=) -
估计步骤 1 中的方差比,该比率将用作步骤 2 的输入 -
在步骤 1 中使用 2 个 CPU (–n 线程) -
a9 和 a10 是分类协变量,将针对不同级别重写 (–qCovarColList)
Rscript step1_fitNULLGLMM.R
--plinkFile=./input/nfam_100_nindep_0_step1_includeMoreRareVariants_poly_22chr
--phenoFile=./input/pheno_1000samples.txt_withdosages_withBothTraitTypes.txt
--phenoCol=y_binary
--covarColList=x1,x2,a9,a10
--qCovarColList=a9,a10
--sampleIDColinphenoFile=IID
--traitType=binary
--outputPrefix=./output/example_binary
--nThreads=2
--IsOverwriteVarianceRatioFile=TRUE
Rscript step2_SPAtests.R
--vcfFile=./input/genotype_100markers.vcf.gz
--vcfFileIndex=./input/genotype_100markers.vcf.gz.csi
--vcfField=GT
--SAIGEOutputFile=./output/genotype_100markers_marker_vcf.txt
--chrom=1
--minMAF=0
--minMAC=20
--GMMATmodelFile=./output/example_binary.rda
--varianceRatioFile=./output/example_binary.varianceRatio.txt
示例 2
-
二元性状(–性状类型=二元) -
使用稀疏 GRM 拟合空模型 (–useSparseGRMtoFitNULL=TRUE, –sparseGRMFile, –sparseGRMSampleIDFile) -
不要在步骤 1 中估计方差比。 -
当使用稀疏 GRM 拟合空模型且不会应用 LOCO 时,仅使用一个 CPU -
在步骤 2 中将 PLINK 输入用于基因型/剂量 -
p 值为 <= pCutoffforFirth 的标记的效应大小将通过 Firth 的偏倚减少逻辑回归 –is_Firth_beta=TRUE 和 –pCutoffforFirth=0.01 来估计(请注意,此选项正在评估中)
Rscript step1_fitNULLGLMM.R
--sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx
--sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt
--useSparseGRMtoFitNULL=TRUE
--phenoFile=./input/pheno_1000samples.txt_withdosages_withBothTraitTypes.txt
--phenoCol=y_binary
--covarColList=x1,x2,a9,a10
--qCovarColList=a9,a10
--sampleIDColinphenoFile=IID
--traitType=binary
--outputPrefix=./output/example_binary_sparseGRM
Rscript step2_SPAtests.R
--bedFile=./input/genotype_100markers.bed
--bimFile=./input/genotype_100markers.bim
--famFile=./input/genotype_100markers.fam
--AlleleOrder=alt-first
--SAIGEOutputFile=./output/genotype_100markers_marker_plink_step1withSparseGRM_Firth.txt
--minMAF=0
--minMAC=20
--GMMATmodelFile=./output/example_binary_sparseGRM.rda
--is_output_moreDetails=TRUE
--sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx
--sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt
--is_Firth_beta=TRUE
--pCutoffforFirth=0.01
示例 3
-
数量特征(–性状类型=定量)和需要逆归一化(–invNormalize=TRUE) -
使用稀疏 GRM 拟合空模型 (–useSparseGRMtoFitNULL=TRUE, –sparseGRMFile, –sparseGRMSampleIDFile) -
在步骤 1 中估计方差比 (–plinkFile=./input/nfam_100_nindep_0_step1_includeMoreRareVariants_poly_22chr) -
当使用稀疏 GRM 拟合空模型且不会应用 LOCO 时,仅使用一个 CPU -
–需要为 VCF 输入指定色度
Rscript step1_fitNULLGLMM.R
--plinkFile=./input/nfam_100_nindep_0_step1_includeMoreRareVariants_poly_22chr
--sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx
--sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt
--useSparseGRMtoFitNULL=TRUE
--phenoFile=./input/pheno_1000samples.txt_withdosages_withBothTraitTypes.txt
--phenoCol=y_quantitative
--covarColList=x1,x2,a9,a10
--qCovarColList=a9,a10
--sampleIDColinphenoFile=IID
--traitType=quantitative
--invNormalize=TRUE
--outputPrefix=./output/example_quantitative_sparseGRM
Rscript step2_SPAtests.R
--vcfFile=./input/genotype_100markers.vcf.gz
--vcfFileIndex=./input/genotype_100markers.vcf.gz.csi
--vcfField=GT
--chrom=1
--SAIGEOutputFile=./output/genotype_100markers_marker_vcf_step1withSparseGRM.txt
--minMAF=0
--minMAC=20
--GMMATmodelFile=./output/example_quantitative_sparseGRM.rda
--is_output_moreDetails=TRUE
--varianceRatioFile=./output/example_quantitative_sparseGRM.varianceRatio.txt
示例 4
-
二元性状(–性状类型=二元) -
使用完整的 GRM 拟合空模型,该 GRM 将使用 plink 文件中的基因型动态计算 (–plinkFile=) -
估计步骤 1 中的方差比,该比率将用作步骤 2 的输入 -
覆盖现有的步骤 1 输出 – Is覆盖方差RatioFile=TRUE -
步骤 2 中标记的 p.值为 ~3.74 x 10^-7
Rscript step1_fitNULLGLMM.R
--plinkFile=./input/nfam_100_nindep_0_step1_includeMoreRareVariants_poly
--phenoFile=./input/Prev_0.1_nfam_1000.pheno_positive_pheno.txt
--phenoCol=y
--covarColList=x1,x2
--sampleIDColinphenoFile=IID
--traitType=binary
--outputPrefix=./output/example_binary_positive_signal
--nThreads=4
--IsOverwriteVarianceRatioFile=TRUE
Rscript step2_SPAtests.R
--vcfFile=./input/nfam_1000_MAF0.2_nMarker1_nseed200.vcf.gz
--vcfFileIndex=./input/nfam_1000_MAF0.2_nMarker1_nseed200.vcf.gz.tbi
--vcfField=GT
--chrom=1
--minMAF=0.0001
--minMAC=1
--GMMATmodelFile=./output/example_binary_positive_signal.rda
--varianceRatioFile=./output/example_binary_positive_signal.varianceRatio.txt
--SAIGEOutputFile=./output/example_binary_positive_signal.assoc.step2.txt
参考
-
1、Home – SAIGE (saigegit.github.io)[13] -
2、GWAS方法 – SAIGE 校正病例对照失衡的广义线性混合模型 – 知乎 (zhihu.com)[14]
参考资料
cget: https://cget.readthedocs.io/en/latest/src/intro.html#installing-cget
[2]savvy: https://github.com/statgen/savvy
[3]cget: https://cget.readthedocs.io/en/latest/src/intro.html#installing-cget
[4]savvy: https://github.com/statgen/savvy
[5][miniconda: https://docs.conda.io/en/latest/miniconda.html
[6]以下是: https://github.com/saigegit/SAIGE/blob/main/conda_env/createCondaEnvSAIGE_steps.txt
[7]r-saige conda 软件包: https://anaconda.org/bioconda/r-saige
[8]问题 #272: https://github.com/weizhouUMICH/SAIGE/issues/272
[9]BGEN: https://bitbucket.org/gavinband/bgen/overview
[10]SAV: https://github.com/statgen/savvy
[11]BGEN: http://www.well.ox.ac.uk/~gav/bgen_format/bgen_format_v1.2.html
[12]SAV: https://github.com/statgen/savvy
[13]Home – SAIGE (saigegit.github.io): https://saigegit.github.io//SAIGE-doc/
[14]GWAS方法 – SAIGE 校正病例对照失衡的广义线性混合模型 – 知乎 (zhihu.com): https://zhuanlan.zhihu.com/p/388615436
欢迎微信交流
小编微信

扫描二维码
获取更多精彩
公众号

本篇文章来源于微信公众号: 微因