元旦快乐!
在生信分析过程中,序列比对占了很大的比重,于是搜索了下相关软件,发现GPU版本的blast或者CPU-GPU混合计算(H-Blast)的软件比较难编译成功,应该是当时的软件编译环境过于老旧,对一些旧的硬件应该还是适用的。但是,依然找到了有最近发表或在活跃更新的软件,分享一下,期待这些软件可以更新并大规模应用!
Chorus:大型数据库异构GPU+CPU多蛋白序列比对搜索
在GPU显存是瓶颈,无法方便扩展的时代,与CPU混合计算就成为了一个比较可行的选择,有一些软件选择这么处理,Chorus就是其中之一,去年2月份发表在Bioinformatics,来看下这个软件。

Chorus是高效的蛋白质-蛋白质局部比对搜索工具,适用于多个查询序列和大型数据库。当一次输入多个查询序列时,比NCBI-BLASTP快300倍,当输入查询序列少于5000个时,可以领先于最先进的数据库-索引方法(比最快的MMseqs2快2.2-27倍,比最快的DIAMOND快1.1-9倍),同时保持低内存占用。
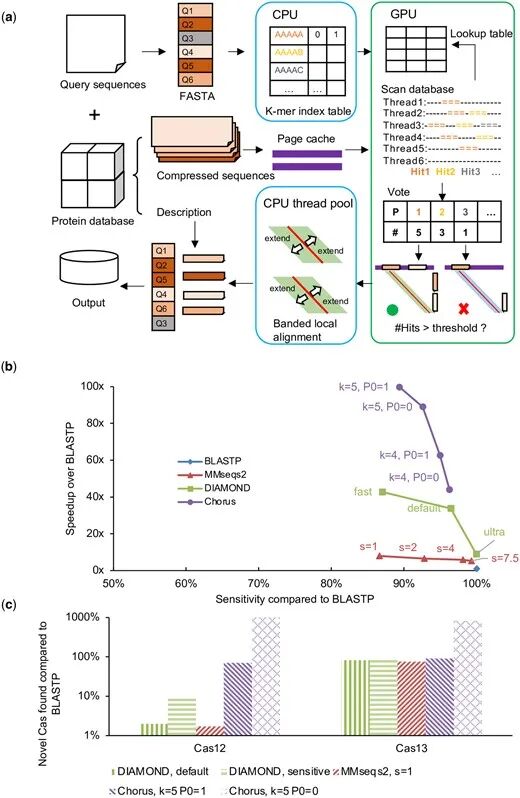
图 软件原理及性能表现
# 软件安装(windows WSL2中进行)
# 英伟达驱动(略)和cuda11.7
wget https://developer.download.nvidia.com/compute/cuda/repos/wsl-ubuntu/x86_64/cuda-wsl-ubuntu.pin
sudo mv cuda-wsl-ubuntu.pin /etc/apt/preferences.d/cuda-repository-pin-600
wget https://developer.download.nvidia.com/compute/cuda/11.7.0/local_installers/cuda-repo-wsl-ubuntu-11-7-local_11.7.0-1_amd64.deb
sudo dpkg -i cuda-repo-wsl-ubuntu-11-7-local_11.7.0-1_amd64.deb
sudo cp /var/cuda-repo-wsl-ubuntu-11-7-local/cuda-*-keyring.gpg /usr/share/keyrings/
sudo apt update
sudo apt-get -y install cuda
# 添加环境变量
export CUDA_HOME=/usr/local/cuda-11.7/
export PATH=$CUDA_HOME/bin:$PATH
export CPATH=$CUDA_HOME/include:$CPATH
export LD_LIBRARY_PATH=$CUDA_HOME/lib64:$LD_LIBRARY_PATH
# 编译,需要cmake
git clone https://github.com/Bio-Acc/Chorus.git
cd Chorus/
mkdir build
cd build
cmake ..cd Chorus/
make
# 然后就获得了createDB和query两个可执行程序
建库程序
./createDB
Usage: <executable_name> <protein_db.fasta> <output_db> [batch_size_GB]
Example: ./creatDB protein_db.fasta output_db 4
参数说明:
-
<protein_db.fasta>:输入FASTA文件,包含蛋白质序列。 -
<output_db>:输出数据库文件的路径。 -
[batch_size_GB]:可选,处理时的批处理大小(GB),默认为GPU显存的三分之一。
查询程序
./query
usage: ./query -q <query.fasta> -d <db1> [db2 ... (optional)] -o <output file> [options]
参数说明:
-
--outfmt INT:输出格式,0为m8表格格式,1为详细比对,2为表格和完整参考序列(fasta格式),3为Opfi格式,4为a3m格式,5为Diamond基准格式。默认为0。 -
-p0, --filter-level INT:通过局部对齐的HSP候选的过滤级别,值越小结果越敏感。默认为1。 -
--min-score INT:显示的每个对齐的最小分数。默认为0。 -
-e, --max-evalue FLOAT:显示的每个比对的最大期望值。默认为10。 -
--max-output-align INT:每个查询显示的最大比对数,0表示无限制。默认为0。 -
--num-threads INT:CPU局部比对的线程数。默认为16。 -
-k, --seed-length INT:k-mer的长度(3, 4, 5)。默认为5。 -
-w, --qit-width INT:查询k-mer的宽度。默认为4。 -
-h, --hash-size INT:哈希表大小比例,值越大GPU投票哈希表越大(每增加一倍)。默认为2。 -
--band-width INT:局部比对的宽度。默认为8。 -
--must-include STRING INT:要求参考序列必须匹配正则表达式N次。例如:--must-include "R[A-Z]{4,7}H" 2。
建库时需要指定batch大小,如没有默认是显存的1/3,建议不要手动将该值增加到GPU内存的一半以上,应该是会显著减慢运行速度。照旧把编译好的二进制文件上传到了百度网盘,回复 “Chorus”自取。软件也有docker版本,可以拉取使用。
MMseq2新增GPU支持:超快速和敏感的序列搜索和聚类软件

这个老牌的软件大家应该是相对熟悉的,github上的星标已经1.5K+,文章引用也有2745次啦!GPU支持刚刚引入,2024.11上传在了预印本,期待早日发表在CNS上。但是软件的设计思路没有采用混合,是纯GPU的,如果数据库大,对显存的要求很高,只能寄望于未来的优化啦!

MMseqs2(多对多序列搜索)用于搜索和聚类大蛋白质和核苷酸序列集,其运行速度是BLAST的10000倍。在其速度的100倍时,它几乎达到了同样的灵敏度。它可以执行profile搜索与PSI-BLAST相同的灵敏度超过400倍的速度。MMseqs2-GPU在NVIDIA L40S GPU上的搜索速度比在128核CPU上的MMseqs2 k-mer快20倍,便宜71倍。软件需要安培或更新架构的NVIDIA GPU才能全速运行,特斯拉一代GPU也可以以低速运行。
# 官方有二进制文件发布,安装就很简单啦,只要驱动够新就行的
# 能在glibc >= 2.29的系统上使用。不需要安装CUDA,只需要nvidia驱动>=525.60.13
wget https://mmseqs.com/latest/mmseqs-linux-gpu.tar.gz
tar xvzf mmseqs-linux-*.tar.gz
# 就可以开心使用啦
MMseqs2 (Many against Many sequence searching) is an open-source software suite for very fast,
parallelized protein sequence searches and clustering of huge protein sequence data sets.
Please cite: M. Steinegger and J. Soding. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nature Biotechnology, doi:10.1038/nbt.3988 (2017).
MMseqs2 Version: 747c64cc8db3b4803a0f1194a3f75b3ba9f81bcb
© Martin Steinegger (martin.steinegger@snu.ac.kr)
usage: mmseqs <command> [<args>]
参数说明:
纯文本输入/输出:
-
easy-search:进行敏感的同源性搜索。 -
easy-cluster:进行慢速但敏感的聚类。 -
easy-linclust:快速线性时间聚类,但敏感度较低。 -
easy-taxonomy:进行分类学分类。 -
easy-rbh:寻找互为最佳命中(Reciprocal Best Hit, RBH)。
数据库输入/输出:
-
search:进行敏感的同源性搜索。 -
map:映射几乎完全相同的序列。 -
rbh:进行互为最佳命中搜索。 -
linclust:快速但敏感度较低的聚类。 -
cluster:进行慢速但敏感的聚类。 -
clusterupdate:用新序列更新之前的聚类。 -
taxonomy:进行分类学分类。
输入数据库创建:
-
databases:列出并下载数据库。 -
createdb:将FASTA/Q文件转换为序列数据库。 -
createindex:存储预计算的索引在磁盘上,以减少搜索开销。 -
convertmsa:将Stockholm/PFAM MSA文件转换为MSA数据库。 -
msa2profile:将MSA数据库转换为profile数据库。
下游处理的格式转换:
-
convertalis:将对齐数据库转换为BLAST-tab、SAM或自定义格式。 -
createtsv:将结果数据库转换为制表符分隔的文件。 -
convert2fasta:将序列数据库转换为FASTA格式。 -
taxonomyreport:以Kraken或Krona格式创建分类报告。
参考:
1.https://github.com/soedinglab/MMseqs2/wiki
2.https://www.biorxiv.org/content/10.1101/2024.11.13.623350v1.full
3.https://github.com/Bio-Acc/Chorus
本篇文章来源于微信公众号:微因