一个可以进行在线HLA imputatation和关联分析的网站,要不要试试呢?有点疑惑的是并没有把HAN.MHC这个参考放上去,毕竟这个有1万人的样本量,应该相对更准确的。
 以下内容转载自官方网站说明:
以下内容转载自官方网站说明:2022年11月2日,《核酸研究》(Nucleic Acids Research)在线发表了复旦大学生命科学学院/人类表型组研究院徐书华教授团队开发的人类主要组织相容性复合体(MHC)基因数据库和计算分析平台PGG.MHC(https://www.pggmhc.org/pggmhc或https://pog.fudan.edu.cn/pggmhc),论文题为“PGG.MHC: toward understanding the diversity of major histocompatibility complexes in human populations”。PGG.MHC收录了来自世界多个族群的53,254个个体的全基因组测序数据(WGS)/全外显子组测序数据(WES)/芯片数据,整合了其MHC基因型和单倍型频率信息,并在此基础上提供了一体化的查询、可视化、数据分析平台。
PGG.MHC数据库共收录了来自世界66个国家的190个族群的53,254个个体的HLA基因型数据,并提供了45,761条基因型频率条目。此外,该工作还首次构建了101个世界人群和33个中国省份人群的HLA单倍型图谱。
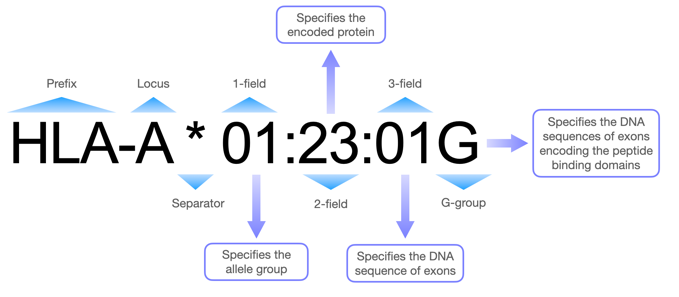
图1 HLA命名原理图
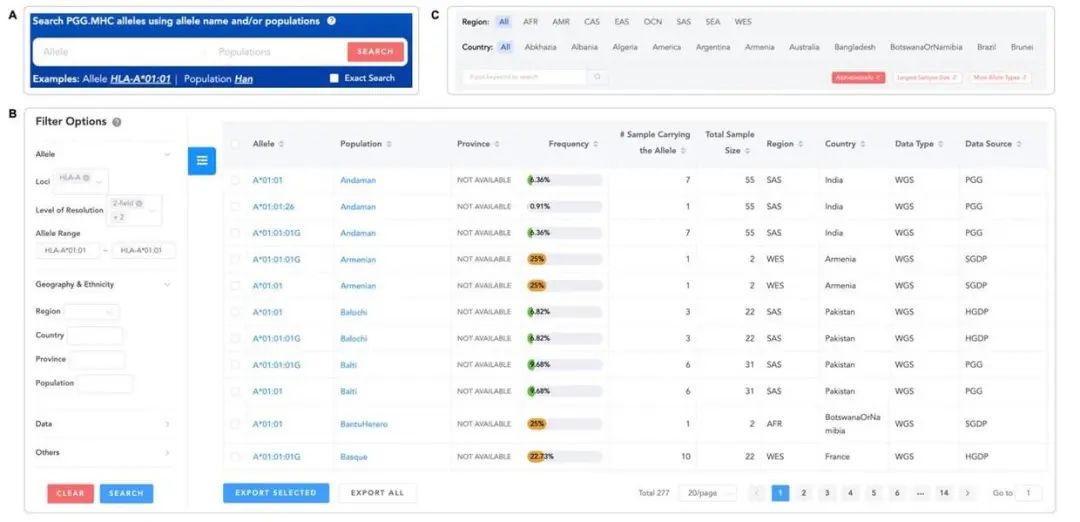
图2 PGG.MHC查询界面示意图
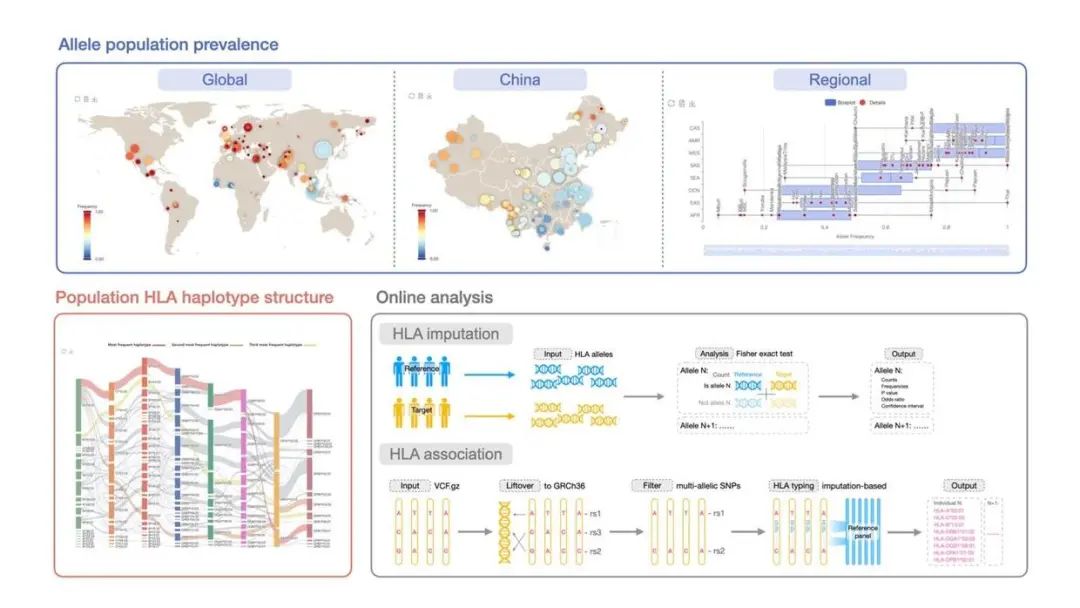
图3 PGG.MHC功能模块
简要地讲,PGG.MHC提供了高质量的HLA基因型和单倍型多样性数据信息,在数据层面对数据来源、HLA分型流程和数据整合各步骤严格把关,保证了频率数据的高可靠性;在数据库功能方面,提供了直观而丰富的数据可视化界面和实用的HLA相关数据分析工具。


关于HAN.MHC参考的使用附如下:
一段时间以来,一直困惑于这个HLA填充参考怎么使用,直到看到了这篇论文课题组的博士论文《中国汉族人白癜风 MHC 区域精细定位研究》。文中较详细描述了怎样使用这个参考,具体过程如下:
1.下载数据集
数据集的原始数据在这里:http://gigadb.org/dataset/100156
只提供了两个文件,感觉和其他的参考数据集的所不同,缺少好几个文件的样子,我也是一直这样困惑,以为作者故意公开信息不全,为了自己的博硕毕业考虑?后来才发现这两个文件已经包含了全部的文件信息。
#下载这两个文件并解压和重命名
wget ftp://parrot.genomics.cn/gigadb/pub/10.5524/100001_101000/100156/HAN.MHC.reference.panel.bgl.gz
gunzip HAN.MHC.reference.panel.bgl.gz
wget ftp://parrot.genomics.cn/gigadb/pub/10.5524/100001_101000/100156/HAN.MHC.reference.panel.markers
2.参考数据集的处理
核心就是这段代码啦,还是挺有用的,自己处理也是可以的,不过有现成的工具就更好啦!
“
将 bgl.phased 文件转换为 PLINK 软件的 ped 和 map 格式数据;然后,再进一步用 PLINK 软件将上述数据转成二进制的 bed、bim 和 fam 格式文件,再计算每个位点的等位基因频率,得到 FRQ.frq 文件。最终得到 SNP2HLA 软件要求的 MHC区 域 参 考 数 据 集 的 所 有 文 件 , 即 含 有 10,689 例 样 本 的 MHC 区 域(chr6:28,477,833~33,448,188bp)29,948 个位点信息的 markers、bgl.phased、bed、bim、fam 和 FRQ.frq 六个文件,其中的位点包括 HLA 等位基因、HLA 基因对应的氨基酸位点、SNPs 和 Indels 四种类型。
#bgl to ped map
#!/usr/bin/perl
use 5.010;
open IN, "$ARGV[0]";
%hash;
@sample;
@sex;
$famid=readline IN;
@famid=split/s+/,$famid;
$nn=1;
$mm=($#famid-1)/2;
for($nn..$mm){
push (@sample, $famid[2*$_+1]);
}
readline IN;
readline IN;
readline IN;
$gender=readline IN;
@gender=split/s+/,$gender;
$nn=1;
$mm=($#gender-1)/2;
for($nn..$mm){
push (@sex, $gender[2*$_+1]);
}
for(0..$#sample){
$sexid{$sample[$_]}="$sex[$_]";
}
@snp=();
while(<IN>){
$line=$_;
@line=split/s+/,$line;
$nn=($#line-1)/2;
push (@snp,$line[1]);
for(1..$nn){
$hash{$famid[$_*2].$line[1]}="$line[$_*2]t$line[$_*2+1]";
}
}
say "@snp";
close IN;
#打印 map 文件
open IB,">map.txt";
foreach my $snp (@snp){
chmop;
my @map = qw (0);
push (@map,$snp,0,0);
say IB "@map";
}
#打印 ped 文件
open IC,">ped.txt";
foreach my $sam (@sample){
chmop;
my @new = ();
push (@new,$sam,$sam,0,0,$sexid{$sam},1);
foreach my $snp (@snp){
chmop;
if(defined($hash{$sam.$snp})){
push (@new,$hash{$sam.$snp});
}else{
$null=null;
push (@new,$null,$null);
}
}
say IC "@new";
}
perl bgl2ped.pl HAN.MHC.reference.panel.bgl 系统资源消耗还是挺大的, 一般的个人电脑是hold不住的,得上服务器。 运行完成后就获得了map.txt和ped.txt两个文件,生成后重命名到你喜欢的名字就好啦!
运行完成后就获得了map.txt和ped.txt两个文件,生成后重命名到你喜欢的名字就好啦!
mv ped.txt ped.ped
mv map.txt ped.map
plink --file ped --make-bed --out HAN.MHC
plink --bfile HAN.MHC --freq --out HAN.MHC
到这里就获得了参考的全部文件啦,下面需要处理的就是处理你的文件以适应这个参考了。根据nature那篇论文,参考是基于hg18的,SNP2HLA只识别SNP名,不管位置的,所以我们只要把SNP名字改成完全一致的就好啦。
3. 调整数据以适应参考
1)坐标转换
一般我们的芯片是基于hg38/hg19的,所以首先就是把参考中的SNP位置转换到我们的位置。Lift Genome Annotations (ucsc.edu)我是用这个网页工具解决的,3万位点左右还是OK的。
2)调整bim文件
这里我用的是自己写的一个简单python脚本解决的,详细如下:
# let snp_name suit for HAN.MHC ref
# get dict
dic = {}
i = 0
maker = ''
with open('dict_hg19-HG18_SY.txt') as f1:
for line in f1:
if i ==0:
i += 1
continue
else:
if line.strip() != "":
marker = line.strip().split('t')[5]
dic[marker] = ''
dic[marker] = line.strip().split('t')[0]
j = 0
fout = open('changed.bim',"w")
with open('8k.bim') as f:
for line in f:
#print(line.strip().split('t')[0])
#break
if line.strip().split('t')[0] !='6':
fout.write(line)
else:
if line.strip() != "":
snp = line.strip().split('t')[1]
if snp in dic.keys():
line = line.strip().split(snp)[0] + dic[snp] + line.strip().split(snp)[1] + 'n'
print(line)
fout.write(line)
#break
j += 1
else:
fout.write(line)
print(j)
fout.close()
一般不填充的芯片大概MHC区域大概有几千位点啦!
4.运行
发现基本上十几个样本要运行8个个小时左右的样子,这里我分配了160G的内存,实际看下来十几个样本需要30多G左右。
nohup ~/biosoft/SNP2HLA_package_v1.0.3/SNP2HLA/SNP2HLA.csh ./hla_16 ~/biodata/HAN.MHC/HAN.MHC 16_results ~/data_home/biosoft/SNP2HLA_package_v1.0.3/SNP2HLA/plink 160000 1000 &
5. 结果整理
这里编写了一个R脚本,方便简单提取结果,未填充成功的,多个结果的,均标记为了NA.
# ########################################
# ####Date 2021.12.25#####################
# ####Author ZJD #########################
# ####Version 1.09########################
# ##SNP2HLA结果计算脚本###################
# ########################################
library(tidyverse)
# 读取填充文件
hla_raw <- read.table('./16_results.bgl.phased', sep = ' ', header=TRUE)
# 预备两个结果文件,选择有用行列
hla.2digits.result <- hla_raw[c(1,4),-c(1,2)]
hla.4digits.result <- hla_raw[c(1,4),-c(1,2)]
# 去除多余行
hla_raw <- hla_raw[-c(1,2,3,4),]
# 找到含有HLA的行
hla_raw.2 <- hla_raw[grepl("*HLA*P*",hla_raw[,2]),]
# 提取基因座名
HLA_name <- hla_raw.2[,2]
# 判断是否有结果
get_na_or_result <- function(result){
if (length(result)>1) {
result <- NA
}
result
}
# 获得一个基因座的2位和4位结果
get_hla_genotype <- function(hla_one_hap, HLA.gene){
HLA.gene.2digits.result <- hla_one_hap[grepl(paste0(HLA.gene,'_[0-9][0-9]$'),
hla_one_hap[,1]),1]
HLA.gene.2digits.result <- get_na_or_result(HLA.gene.2digits.result)
if (length(HLA.gene.2digits.result)==0) {
HLA.gene.2digits.result=NA
}
HLA.gene.4digits.result <- hla_one_hap[grepl(paste0(HLA.gene,
'_[0-9][0-9][0-9][0-9]$'),
hla_one_hap[,1]),1]
HLA.gene.4digits.result <- get_na_or_result(HLA.gene.4digits.result)
if (length(HLA.gene.4digits.result)==0) {
HLA.gene.4digits.result=NA
}
HLA.one_gene.result <- list(HLA.gene.2digits.result=HLA.gene.2digits.result,
HLA.gene.4digits.result=HLA.gene.4digits.result)
}
# 主程序,获得每个样本每个基因的分型
for (s in colnames(hla_result)[-c(1,2)]) {
# 获得每个样本的结果
hla_one_hap <- hla_raw.2[grepl("P",hla_raw.2[,s]),c("pedigree",s)]
hla.gene.li <- c('HLA_A', 'HLA_C', 'HLA_B', 'HLA_DRB1', 'HLA_DQA1',
'HLA_DQB1', 'HLA_DPA1', 'HLA_DPB1')
for (HLA.gene in hla.gene.li) {
HLA.one_gene.result <- get_hla_genotype(hla_one_hap, HLA.gene)
hla.2digits.result[HLA.gene,s] <- HLA.one_gene.result$HLA.gene.2digits.result
hla.4digits.result[HLA.gene,s] <- HLA.one_gene.result$HLA.gene.4digits.result
}
}
# 文件写出
write.table(hla.2digits.result, 'hla.2digits.result.txt', quote=FALSE)
write.table(hla.4digits.result, 'hla.4digits.result.txt', quote=FALSE)
本篇文章来源于微信公众号: 微因