最近听了菲沙基因的网课,记录一下!多数是其课程ppt的截图,如有侵权,立马删除。声明,和这个公司无利益相关,只是为了学习和分享知识。
群体遗传分析步步骤总览
主要包括7个步骤,分别是:SNP过滤、基因型填充、进化树分析、主成分分析、群体遗传结构分析、连锁不平衡分析和GWAS分析。

1.SNP过滤

主要使用vcftools进行,所以输入文件是测序后分析出的vcf文件,生成的还是vcf。
软件主页:http://vcftools.github.io/
vcftools主要用于处理vcf文件,功能有以下几个:
- 过滤特定变异
- 比较文件
- 汇总变异
- 转换成其他文件格式
- 验证和合并文件
- 创建交集和子集

过滤连锁不平衡,使用plink进行:
软件主页:http://zzz.bwh.harvard.edu/plink/

2.基因型填充
使用beagle软件进行,输入输出也都是vcf文件。java软件包,先要有java环境。
软件主页:http://faculty.washington.edu/browning/beagle/beagle.html
wget http://faculty.washington.edu/browning/beagle/beagle.18May20.d20.jar
mv beagle.18May20.d20.jar beagle.jar

3.进化树构建

使用Mega软件进行,这个软件大家都熟悉吧,各个平台都有,但是用下来好像只有windows版本的比较好用,mac的基本上是废的,动一下就卡,应该是采用wine之类的模拟搞的。linux估计也是如此。ppt上的步骤已经很详细了,直接截个图了。

4. PCA

使用GCTA软件进行的,bioconda直接安装就行了。
conda install -c biobuilds gcta
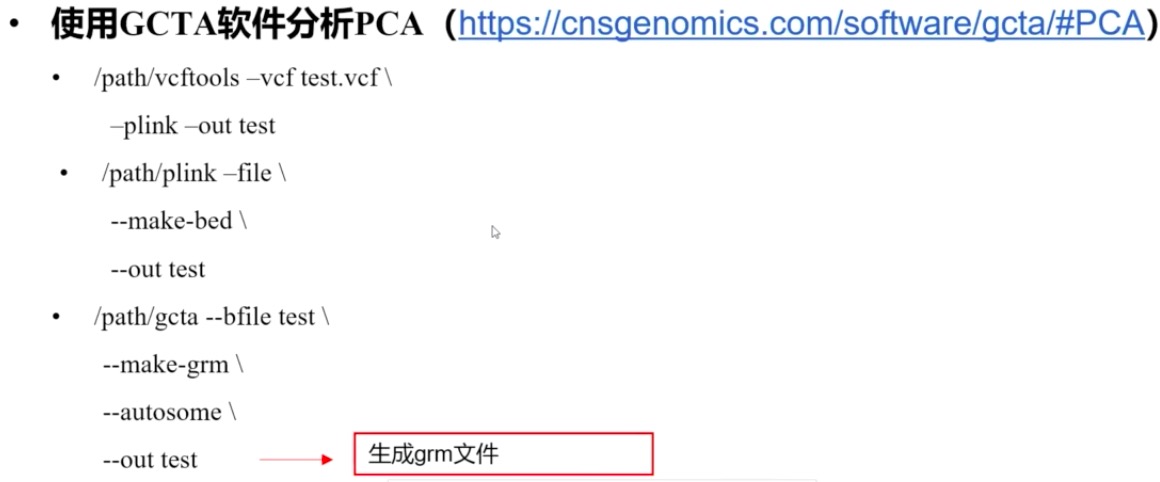
构建G矩阵(GWAS数据中个体间遗传关系的估计),会生成三个grm文件,分别是grm.bin, grm.N.bin和grm.id,分别是下面的含义:
运行后,会得到如下四个文件:
- test.grm.bin 含G阵下三角元素,是二进制文件
- test.grm.N.bin 记录计算G阵的SNP个数,是二进制文件
- test.grm.id 记录个体的family号和id号,即plink fam文件的前两列
- kinship.log 日志文件。
5.群体结构分析
vcftools转换格式从vcf到plink格式ped,然后plink转换成二进制格式bed。
admixture进行群体结构分析。
#软件可以使用conda进行软件安装
conda install admixture

6.连锁不平衡分析

7.GWAS
一个R包,第一次听说,官网:http://www.zzlab.net/GAPIT/

这是需要的两个矩阵,听课程中说是可选的。


从网址看是华大基因开源的一个画图的R包,主要是画曼哈顿图,因图像纽约曼哈顿区的摩天大楼而得名。

到这里,一个流程就结束了,是不是想摩拳擦掌实践一下了?欢迎交流呀!