接着前面的内容,这里再进行下数据库的处理,看看从参考数据库就按测序数据处理是不是能提高物种注释的精度。这里先预报一下,种的分类结果并不能有明显的提升,或许是因为序列长度的缺陷,即使再努力提高技巧,终究不能解决根本的问题,250bp的长度,对比1500bp左右的全长,显然还是太短了,难以实现精确的分类,所以,要想更精确,只有上16S全长,这只能寄望于Pacbio,Oxford Nanopore,和10x linked reads或者类似的技术,比如华大的sLtFR等技术提升读长了。再激进些,等测序成本足够低,上宏基因组,宏转录组了。
Anyway,记录一下我这次探索的过程。
1.使用qiime2截取V4区域
其实,自己写脚本也是能搞定的,但是,毕竟自己的水平自己清楚哈,能用大神的就用大神的。大神经验丰富,能够解决引物配的问题,比如两个碱基模糊匹配,以模拟真实PCR过程中的错配情况等等。要我做的话,只能简单正则表达式解决完全匹配的情况,注定要miss一些序列,这会对准确性造成一定影响,特别是碰巧这些序列是你需要的话。以下是我的步骤:
#激活conda和进入qiime环境,如果你需要conda推荐这篇[1]
source ~/Miniconda3/bin/activate
conda activate qiime2-2019.10
#导入Silva132数据库中的原始序列(99%相似度聚类)
#下载自这里https://www.arb-silva.de/fileadmin/silva_databases/qiime/Silva_132_release.zip
qiime tools import \
--type 'FeatureData[Sequence]' \
--input-path SILVA_132_QIIME_release/rep_set/rep_set_16S_only/silva_132_99_16S.fna \
--output-path s_otus.qza
##截取V4区域,使用515F-806R, V4最新引物,emp 16s protocol[2]
time qiime feature-classifier extract-reads \
--i-sequences s99_otus.qza \
--p-f-primer GTGYCAGCMGCCGCGGTAA \
--p-r-primer GGACTACNVGGGTWTCTAAT \
--o-reads sref-seqs.qza
2.序列探索和处理
首先,把序列提取出来,qiime2的序列qza文件是可以直接重命名为zip格式文件解压的,就解压sref-seqs.qza后从文件夹里的data子文件夹里找到了序列文件,dna-sequences.fasta,368240条序列。
稍微统计了一下序列长度分布,以及在其中找不找得到引物。发现只有少部分的序列能够找到引物,一般是只能找到一端,应该讲,软件是可以直接切去引物的,留下的应该只有v4区的真正序列而已。而这少部分能找到引物的序列,应该是在其他位置错误匹配到了引物序列。中间有个小插曲,最后一个序列始终会丢掉,由于几个月没怎么用python,算法有点生疏了,于是只能在获得的序列直接加了个随便的序列名暂时解决了。用了个脚本来解决:
def deal_with_seq(seq,seq_name):
'''
简单的以引物中不存在简并的几个碱基来查找序列中引物存在与否,以及其位置
然后,处理这些序列为120+10+120
'''
primer_F = 'GCCGCGGTAA'
primer_R = 'ATTAGA'
seq_F = ''
seq_R = ''
if primer_F in seq and primer_R not in seq: #只找到正向引物的情况
seq_F = seq.split(primer_F)[1][1:120] #正向引物后120bp,对应之前的测序reads切到120bp
seq_R = seq.split(primer_F)[1][-120:] #同上
print(len(seq.split(primer_F)[1])) #看下除了引物后的序列长度
seq = seq_F + "NNNNNNNNNN" + seq_R #以10个N连接,对应之前的数据处理
elif primer_R in seq and primer_F not in seq: #只找到反向引物的情况
seq_R = seq.split(primer_R)[0][-120:]
seq_F = seq.split(primer_R)[0][:120]
#print(seq_F, seq_R)
print(len(seq.split(primer_R)[0]))
seq = seq_F + "NNNNNNNNNN" + seq_R
elif primer_F in seq and primer_R in seq: #双向引物都找到的情况
print(len(seq.strip()))
seq_F = seq.split(primer_F)[1][:120]
seq_R = seq.split(primer_R)[0][-120:]
seq = seq_F + "NNNNNNNNNN" + seq_R
else: #其他的情况,默认是只有v4区序列了
print(len(seq.strip()))
seq = seq[:120] + "NNNNNNNNNN" + seq[-120:]
return seq
def get_250_otus():
'''
读取和存储这些序列
'''
fout = open('250_otus.fasta', 'w')
f_length = open('250_otus_length.txt', 'w')
f_undealed = open('undealed.fasta', 'w')
dic_length = {}
with open('dna-sequences.fasta') as f:
seq_name = ''
seq = ''
for line in f:
if line.startswith('>'):
if seq_name != '':
dic_length[seq_name] = 0
dic_length[seq_name] = len(seq)
seq = deal_with_seq(seq, seq_name)
f_undealed.write(seq_name + seq + '\n')
fout.write(seq_name + seq + '\n')
f_length.write(seq_name + str(dic_length[seq_name]) + '\n')
seq_name = line
seq = ''
else:
seq_name = line
elif line.strip() != '':
seq += line.strip()
fout.close()
f_length.close()
f_undealed.close()
if __name__ == '__main__':
get_250_otus()
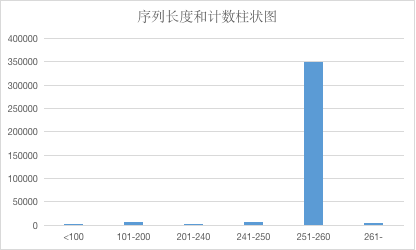
统计一下序列长度分布:
从柱状图可以看出,绝大绝大多数是251-260bp的长度,其余的序列是可以忽略的,这个结果还是比较靠谱的。
3.训练模型,测试物种注释效果
告别前面自己造的小轮子,回归qiime2的流程。
#重新导入序列
qiime tools import \
--type 'FeatureData[Sequence]' \
--input-path /Users/zd200572/Biodata/Refrence/8c74b73b-c49b-44f1-bfba-54df23b1c8d3/data/250_otus.fasta \
--output-path s250_otus.qza
#训练模型,比较在耗时,特别是我的笔记本性能一般,大概相当于i5一代的水平,16G内存
time qiime feature-classifier fit-classifier-naive-bayes \
--i-reference-reads s250_otus.qza \
--i-reference-taxonomy sref-taxonomy.qza \
--o-classifier classifier_silva_V4.qza
#两个多小时,8140.54s user 1138.65s system 93% cpu 2:45:32.42 total
#物种注释
time qiime feature-classifier classify-sklearn \
--i-classifier classifier_silva_V4.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy250.qza
#754.47s user 57.61s system 80% cpu 16:47.93 total
4.和之前的模型的比较
在其他分类级别上没有明显差别,在属上有明显地提升从194->283,看了一下基本上分类明确的属,做的这一切还是比较值得的。但是还要确定是否有假阳性的情况,种上的分类情况就不忍直视了,基本上是未分类种。
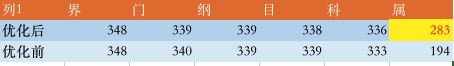
再来看看优化后属的柱状图,分类确实更好了:

参考: