简介
picrust2 beta既可以单独安装,也可以以qiime2-PICRUST2插件方式安装和使用,两者都可以在linux和Mac上运行,windows请使用虚拟机。
请注意,与PICRUSt1元基因组预测步骤相比,使用默认流程和ASV并使用PICRUSt2管道生成分层的输出表(通过贡献序列分解的预测)需要更多的计算资源。至少需要16GB的内存,几个小时(取决于你的CPU核心数和输入样本数目)。如果使用非默认方法将ASV放入参考树中,则内存需求较低(本教程中对此进行了介绍)。官方推荐使用conda方式进行安装:
#安装好conda后使用下面命令新建一个环境
conda create -n picrust2 -c bioconda -c conda-forge picrust2=2.2.0_b
conda activate picrust2
基本的qiime2-PICRUST2插件可以从官网找到安装方式,这个插件可以嵌入qiime2的分析流程中,使用降噪方法而不是closed-reference OTU聚类。安装方法可以在官网找到https://library.qiime2.org/plugins/q2-picrust2/13/。其实就一个命令,附在这了:
#最新版本是2019.7,10月版本还未发布,我试下与10月版本的兼容性
conda activate qiime2-2019.7
conda install q2-picrust2=2019.7 -c conda-forge -c bioconda -c gavinmdouglas
在开始这个教程之前,我们建议你浏览独立教程,以更好地了解该工具。许多独立版本的特性在插件版本中还不能用,你还应该读下宏基因组预测的关键局限性。
运行细节
运行qiime2-PICRUST2流程至少需要16GB的内存(或许更多)。这比PICRUST1提高了许多,因为不再为特定的16S rRNA基因数据库预先计算预测, 而是对每个数据集运行基因组预测步骤。
在q2-picrust2中有两个用于运行PICRUSt2的流程:
- 1.涉及使用EPA-NG运行序列放置的默认流程。 下面的教程将重点介绍这种方法。
- 2.修改后的流程从另一个工具读取已放置的序列(例如q2-fragment-insertion)。 为了避免增加的内存需求,您可以使用此流程来使用替代的读取放置方法,这通常是PICRUSt2占用内存最多的步骤。 有关详细信息,请参见此处。将与q2-fragment-insertion一起放置的ASV用作q2-picrust2插件的输入。
运行示例
你可以使用如下命令获得PICRUST2帮助信息:
qiime picrust2 full-pipeline --help
所需的输入是–i-table和–i-seq,它们分别需要对应于FeatureTable [Frequency]和FeatureData [Sequence]类型的QIIME2文件。 特征表需要包含大量的ASV(即BIOM表),并且序列文件必须是每个ASV序列的FASTA文件。 独立的PICRUSt2版本有很多选项,但目前仅选择线程数(–p线程),隐藏状态预测(HSP)方法(–p-hsp-method)和最大NSTI值( –p-max-nsti)。
–p-max-nsti选项指定在排除参考序列之前,序列在参考系统发育中需要的距离截止值,默认截止值为2。在用于测试PICRUSt2的人类数据集中,唯一超出此默认截止值的ASV在16S数据集中错误地是18S序列,这表明此截止值非常宽松。 对于环境数据集,基于此默认临界值,可能会丢弃更高比例的ASV(Douglas等,准备中)。
我们将使用下面的测试文件,你可以使用如下命令下载:
```bash
mkdir q2-picrust2_test_full
cd q2-picrust2_test_full
wget http://kronos.pharmacology.dal.ca/public_files/tutorial_datasets/picrust2_tutorial_files/mammal_biom.qza
wget http://kronos.pharmacology.dal.ca/public_files/tutorial_datasets/picrust2_tutorial_files/mammal_seqs.qza
wget http://kronos.pharmacology.dal.ca/public_files/tutorial_datasets/picrust2_tutorial_files/mammal_metadata.tsv
这些文件对应于样本的ASV计数表,ASV序列和元数据。 共有11个样本和371个ASV。 这些样品是从北极狼,土狼,海狸和豪猪的哺乳动物粪便中收集的,如先前所述。
你可以用下面命令运行全部的PICRUSt2流程(会花费大约 ~25 min和14 GB RAM – 如果设置更多线程将会更快)。
qiime picrust2 full-pipeline \
--i-table mammal_biom.qza \
--i-seq mammal_seqs.qza \
--output-dir q2-picrust2_output \
--p-threads 1 \
--p-hsp-method pic \
--p-max-nsti 2 \
--verbose
注意在这种情况下,由于它是最快的,所以指示了系统发育独立对比隐藏状态预测方法。 但是,我们建议用户在实践中使用mp方法(此示例在1个线程上约54分钟)。
输出文件像下图的红色框所求示:
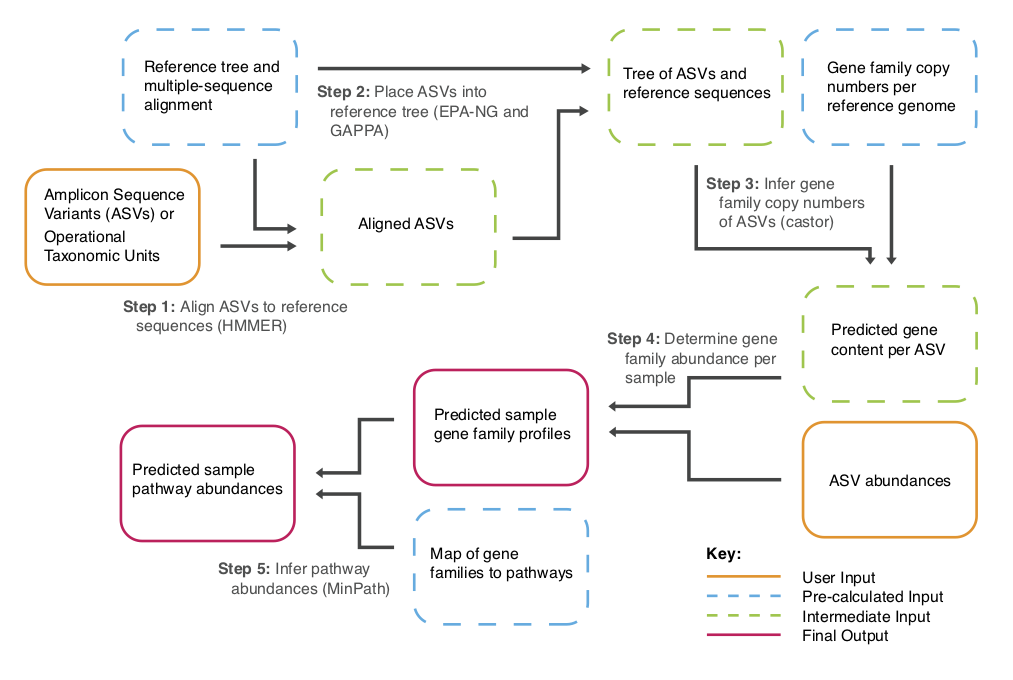
它们分别是(q2-picrust2_output):
ec_metagenome.qza– 预测的宏基因组 EC 数量 (行是 EC数量,列是样本情况).ko_metagenome.qza– 宏基因组KO 预测文件KO (行是 KOs ,列是样本情况).pathway_abundance.qza– MetaCyc 代谢途径相对丰度预测 (行是pathways,列是样本情况).
元件都是FeatureTable[Frequency]`类型的, 也就是说可以使用QIIME2插件处理和分析。
例如,你可以使用如下命令获得样本丰度的汇总信息并可视化。
qiime feature-table summarize \
--i-table q2-picrust2_output/pathway_abundance.qza \
--o-visualization q2-picrust2_output/pathway_abundance.qzv
你可以看到一个表格,类似下表,描述了总的代谢途径相对丰度和各样本的代谢途径相对丰度。下图为qiime2 2019.7版本生成,可能 与之前版本的结果有细微差别。注意这不是最终结果,最终应该是上面创建的FeatureTable[Frequency]` 。
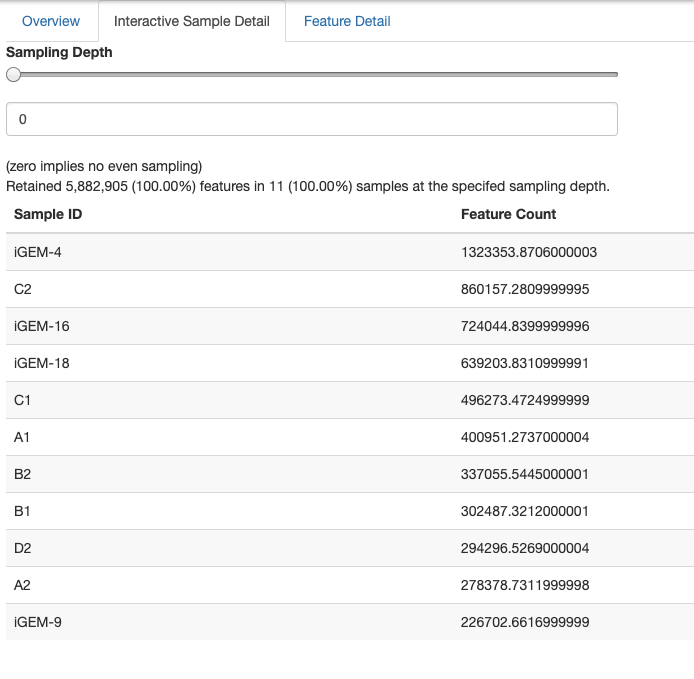
注意这个文件不是以相对丰度为单位,而是每个ASV贡献的预测功能丰度的总和乘以每个ASV的丰度(输入读取的数目)。
上面的宏基因组预测可以嵌入到几个QIIME2分析过程中,例如,你可以基于这些表,快速计算多样性信息。样本最小代谢途径丰度为226702,所以我们,因此我们在计算核心多样性指标时会稀疏到此临界值:
qiime diversity core-metrics \
--i-table q2-picrust2_output/pathway_abundance.qza \
--p-sampling-depth 226702 \
--m-metadata-file mammal_metadata.tsv \
--output-dir pathabun_core_metrics_out \
--p-n-jobs 1
看一下pathabun_core_metrics_out/bray_curtis_emperor.qzv` ,你应该会获得一个类似下图的图,您应该看到这样的排序图,它显示了食肉动物和啮齿动物之间该数据集之间的明显差异:
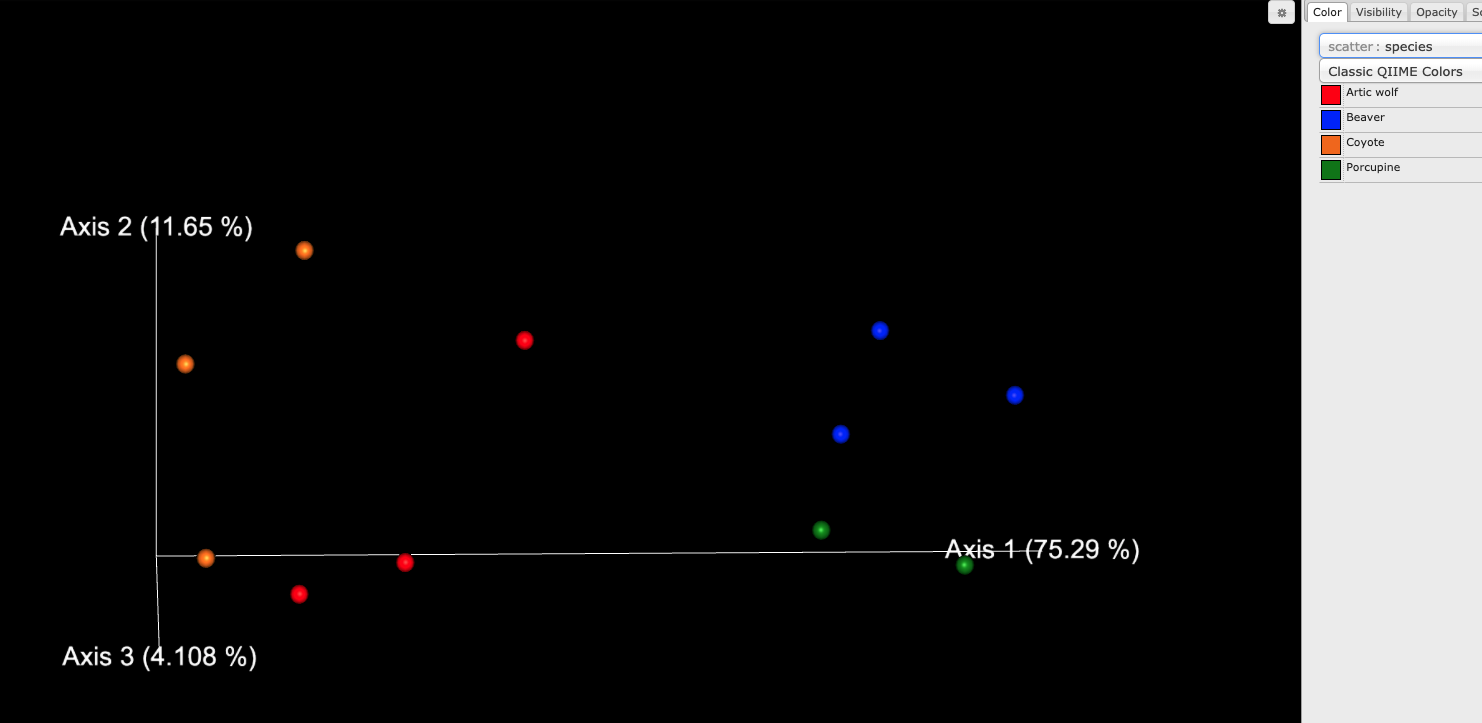
最后输出的QZA的文件是 QIIME2 FeatureTable[Frequency],可以在QIIME2中使用。 如果你想在其他软件中使用,用下面命令转换成BIOM格式:
qiime tools export \
--input-path q2-picrust2_output/pathway_abundance.qza \
--output-path pathabun_exported
下面这个命令将BIOM文件转换成一个纯文本文件:
biom convert \
-i pathabun_exported/feature-table.biom \
-o pathabun_exported/feature-table.biom.tsv \
--to-tsv
还找到简书上一个使用原来的closed-reference OTU聚类方式的,估计也是翻译自官方,附在这https://www.jianshu.com/p/6a0b85b96874