1. qiime2数据导出
发现自己还是比较菜的,不能在qiime2的结果文件里进行较好的操作来达到获得各种属相对丰度的表,于是决定把数据导出到biom和txt格式来进行后续操作。这样就可以利用qiime2的新算法的优势,和之前已有的宏基因组公众号上介绍的流程来进行后续操作。毕竟,对于16S扩增子分析最重要的就是获得物种分类丰度信息。
qiime tools export \
--input-path feature-table.qza \
--output-path exported-feature-table
2.python脚本处理可视化导出的文件
获得的文件是一个序列计数,没有转化成频率,发现不是怎么好用,于是自写python脚本解决一下:
fout = open('silva_120_freq.csv', 'w')
filein = 'gut_repeat_120_silva_level-7.csv'
lis = []
def get_feature_table_freq(filein):
with open(filein) as f:
for line in f:
line1 = ''
lis = []
lis_freq = []
if line.strip().split(',')[0] == 'index':
lis = line.strip().split(',')
for c in lis: #处理第一行的菌属种名,去除多余的信息,使简洁并且好看
if c == 'index':
line1 += 'index,'
elif ';D_5__' in c:
line1 += 'D_5__' + c.split(';D_5__')[1] + ','
elif c != '' and ';__' in c:
line1 += c.split(';__')[0].split(';')[-1] + ','
line1 += '\n'
#print(line1)print(line1)
fout.write(line1)
else:#处理数据信息,求和后算比例,使用两个列表解决,肯定不是最好的方式,但是是我最容易想到的,矩阵什么的还hold不住。
if line.strip() != '':
sample_name = line.strip().split(',')[0]
lis = line.strip().split(',')[1:]
sum_lis = 0.0
for a in lis[:-1]:
if a != '':
sum_lis += float(a)
line1 += line.strip().split(',')[0] + ','
for b in lis:
if b != '':
#lis_freq.append(float(b)/sum_lis)
line1 += str(float(b)/sum_lis) + ','
line1 += lis[-1] + '\n'
fout.write(line1)
get_feature_table_freq(filein)
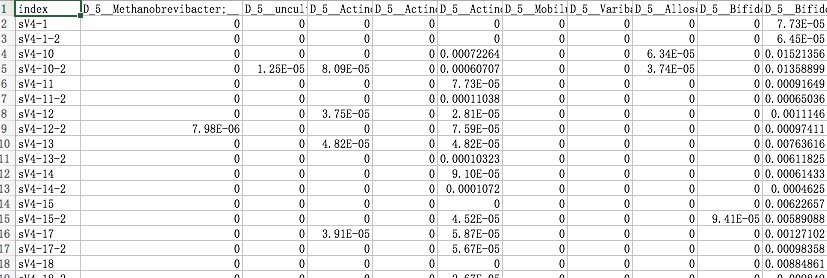
得出的结果符合预期,暂时先这么用着,在没有找到更好的方法之前。
处理后的结果如下图: