肠型,Enterotype,是2011年在这篇文章中提出的,即将过去的2018年又有20多们肠道微生物的大佬对肠型的概念进行了回顾和确认。一直比较好奇怎样来用代码分析肠型,今天找到了这个教程,放在这:
这是那篇原始的文章:Arumugam, M., Raes, J., et al. (2011) Enterotypes of the human gut microbiome, Nature,doi://10.1038/nature09944
在谷歌上一搜,作者竟然做了个分析肠型的教程在这,学习一下:http://enterotyping.embl.de/enterotypes.html
这是2018年大佬们的共识文章:这是国人翻译的这篇文章,http://blog.sciencenet.cn/blog-3334560-1096828.html
当然,如果你只需要获得自己的结果或者自己课题的结果,不需要跑代码的,有最新的网页版分型,更好用,网址也放在这,同样也是上面翻译的那篇文章里提到的网址:http://enterotypes.org/ 只需要把菌属的含量比例文件上就能很快得到结果。
下面我就边学习边做来尝试着来个分析,并把代码放在这里备忘。其实作者已经整理好了代码,我学习一下,争取实现对手上的数据进行分析。
首先下载测试数据,
wget http://enterotyping.embl.de/MetaHIT_SangerSamples.genus.txt
wget http://enterotyping.embl.de/enterotypes_tutorial.sanger.R
跑跑示例数据,排排错
我表示对R语言还只是一知半解的状态,所以,先跑下,然后能用上自己的数据, 当个工具用就暂知足啦。我是黑苹果10.11的系统,运行这个软件提示少了Xquartz,于是装了个,windows和linux应该不需要。原代码中还提示『没有”s.class”这个函数』,百度了一下发现有个老兄的新浪博客说了是这个包,于是加了句library(ade4)就ok了。
Xquartz的下载地址Mac 10.6+:https://dl.bintray.com/xquartz/downloads/XQuartz-2.7.11.dmg
#Uncomment next two lines if R packages are already installed
#install.packages("cluster")
#install.packages("clusterSim")
library(cluster)
library(clusterSim)
#BiocManager::install("genefilter")
library(ade4)
#Download the example data and set the working directory
#setwd('<path_to_working_directory>')
data=read.table("../MetaHIT_SangerSamples.genus.txt", header=T, row.names=1, dec=".", sep="\t")
data=data[-1,]
dist.JSD <- function(inMatrix, pseudocount=0.000001, ...) {
KLD <- function(x,y) sum(x *log(x/y))
JSD<- function(x,y) sqrt(0.5 * KLD(x, (x+y)/2) + 0.5 * KLD(y, (x+y)/2))
matrixColSize <- length(colnames(inMatrix))
matrixRowSize <- length(rownames(inMatrix))
colnames <- colnames(inMatrix)
resultsMatrix <- matrix(0, matrixColSize, matrixColSize)
inMatrix = apply(inMatrix,1:2,function(x) ifelse (x==0,pseudocount,x))
for(i in 1:matrixColSize) {
for(j in 1:matrixColSize) {
resultsMatrix[i,j]=JSD(as.vector(inMatrix[,i]),
as.vector(inMatrix[,j]))
}
}
colnames -> colnames(resultsMatrix) -> rownames(resultsMatrix)
as.dist(resultsMatrix)->resultsMatrix
attr(resultsMatrix, "method") <- "dist"
return(resultsMatrix)
}
data.dist=dist.JSD(data)
pam.clustering=function(x,k) { # x is a distance matrix and k the number of clusters
require(cluster)
cluster = as.vector(pam(as.dist(x), k, diss=TRUE)$clustering)
return(cluster)
}
data.cluster=pam.clustering(data.dist, k=3)
require(clusterSim)
nclusters = index.G1(t(data), data.cluster, d = data.dist, centrotypes = "medoids")
nclusters=NULL
for (k in 1:20) {
if (k==1) {
nclusters[k]=NA
} else {
data.cluster_temp=pam.clustering(data.dist, k)
nclusters[k]=index.G1(t(data),data.cluster_temp, d = data.dist,
centrotypes = "medoids")
}
}
plot(nclusters, type="h", xlab="k clusters", ylab="CH index",main="Optimal number of clusters")
obs.silhouette=mean(silhouette(data.cluster, data.dist)[,3])
cat(obs.silhouette) #0.1899451
#data=noise.removal(data, percent=0.01)
## plot 1
obs.pca=dudi.pca(data.frame(t(data)), scannf=F, nf=10)
obs.bet=bca(obs.pca, fac=as.factor(data.cluster), scannf=F, nf=k-1)
dev.new()
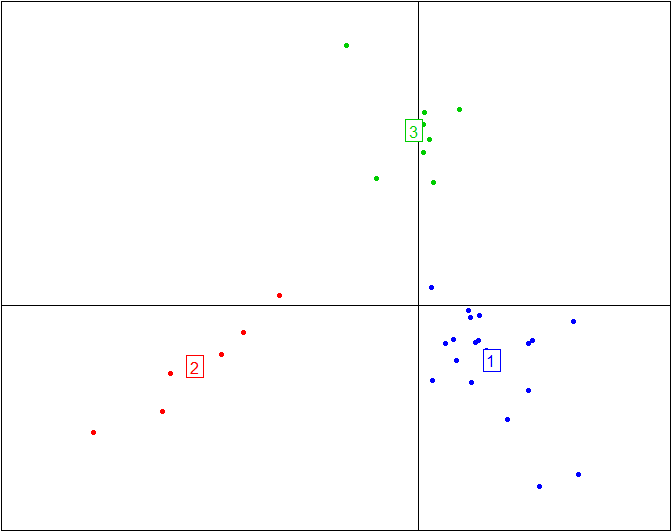
s.class(obs.bet$ls, fac=as.factor(data.cluster), grid=F,sub="Between-class analysis", col=c(3,2,4))
#plot 2
obs.pcoa=dudi.pco(data.dist, scannf=F, nf=3)
dev.new()
s.class(obs.pcoa$li, fac=as.factor(data.cluster), grid=F,sub="Principal coordiante analysis", col=c(3,2,4))
上图,稍微调整下
, col=c(3,2,4)这个代码是给三个聚类上不同的颜色,还没搞清楚怎么给画的圈上色来实现理好看的效果,相信对于熟悉R语言的同学是小菜一碟。, cell=0, cstar=0是不显示圈和边线,只显示散点。
不加这两个参数,只用上面的代码,图如下:
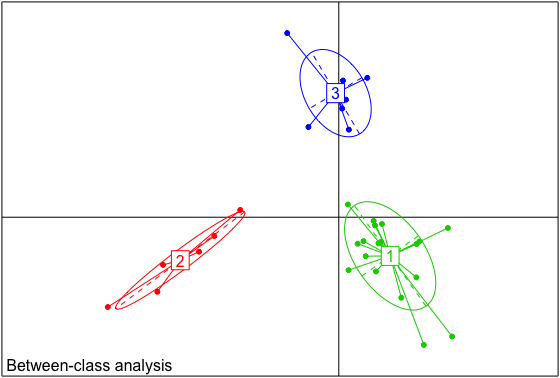
加上两个参数的图片,就和教程里的最后面的两张图一样: