发现一个中国疾控中心的工作人员zhangwen2001做的一个docker,刚好最近在分析肠道微生物的数据,学习学习。动用我的自以为比较厉害的搜索和查找信息的能力,终于找到了镜像文件。先把github的地址放在这:
https://github.com/zhangwen2001/Guthealthy
这个代码库并没有说明docker hub中有这个镜像,但是我怀着侥幸心理搜索了一下,是可以找到的,开心呀!作者的邮箱是zhangwen@icdc.cn,试了一下,icdc.cn网站是中国疾控的,所以我判断作者是这个单位的人。
一、镜像获取
不过pull这个镜像的过程中还是遇到了一点小问题,提示没有默认的latest版本,于是我尝试使用docker hub上标明的v1.0,成功地拉取了下来,当然,docker安装也是有点小费劲,如果有报错的话,还好有官方教程在。
docker pull zhangwen2001/guthealthy:v1.0
二、镜像使用
sudo docker run -it --rm -v /home/zd/microbiome_gut:/test zhangwen2001/guthealthy:v1.0 cd /test perl /bioapp/bin/Guthealthy.pl /test/input.fq /test
得到的报告大概就和软件仓库上介绍的差不多,可以获得的信息有:
1.汇总信息
包括总的reads数,有多少个属,香农指数
2.核心菌群信息
各个菌属的含量,以及正常人的菌群含量平均值
3.致病菌的情况
总共分为三组,从未在正常人中发现的菌;从未在HMP计划健康人中发现的菌;以及其他可能的致病菌。
三、镜像流程初步学习
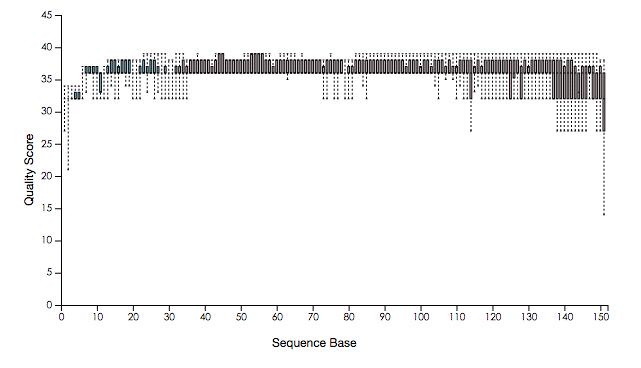
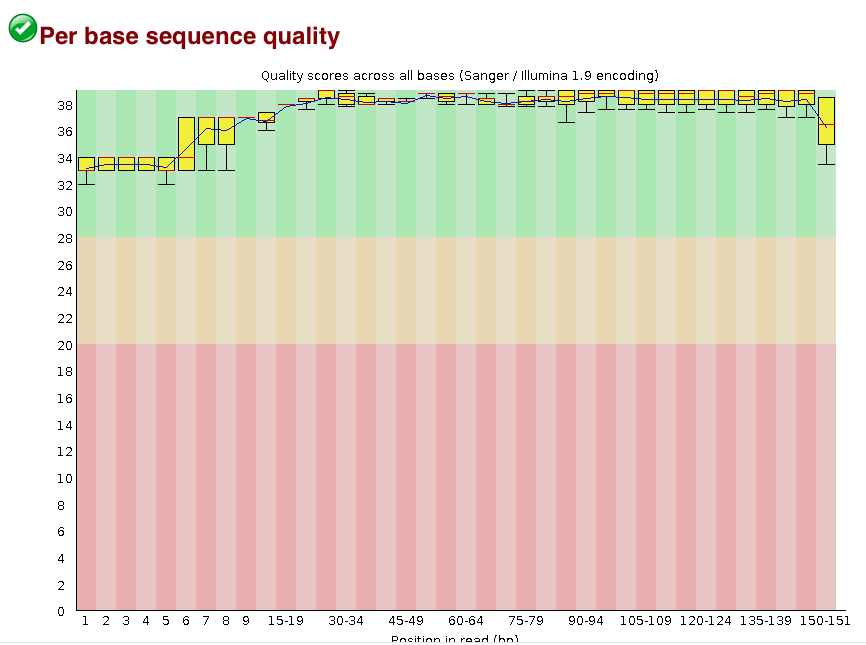
从运行命令来看,这是一个perl脚本,打开来看了一下发现基本上是把各个软件的串联起来,对于我这个perl门外汉也能看懂,窃喜。作者的代码已经注释的相当清楚,第一步,质控,运行了自己的脚本去过滤低质量reads,输出文件里还有一个fastqc的结果,从序列质量图来看,质控做得相当漂亮,有图为证。
质控之前的质量分布图:
质控之后的序列质量分布图:
第二步,统计细菌的多样性,作者没有使用最广泛使用的qiime、mothur、vsearch等软件,而是使用了bowtie2去比对一个自建的参考序列集。然后后面同样用bowtie2获得了致病菌的相关信息。话说我也曾经想过用比对去解决双向测序长度短序列不能较好拼接的问题,苦于没有思路,作者的思路值得我深入学习。最后一步是用一个cat命令生成了报告。
#!/usr/bin/perl use strict; use warnings; my $file=$ARGV[0]; #fq文件 my $out=$ARGV[1]; print "Step 1: Quality Control for Sequence Data 测序数据质量控制\n"; system "perl /bioapp/bin/read_filter_QC.pl -input $file -outdir $out\n"; print "Step 2: Genus Num 样本中菌属多样性\n"; system "/bioapp/bin/bowtie2-2.3.2/bowtie2 -x /bioapp/Data/16S-complete-clean.fa -q $out/filter.fq -S filter.sam\n"; system "perl /bioapp/bin/bowtie_stat-v3.pl filter.sam /bioapp/Data/16S-complete-clean.taxon $out/filter.fq /bioapp/Data/Core_genus filter.stat\n"; print "Step 3:Pathogen Detection 样本中含有的病原菌\n"; system "/bioapp/bin/bowtie2-2.3.2/bowtie2 -x /bioapp/Data/155pathogens.fa -q $out/filter.fq -S filter.bwa.sam\n"; #system "/bioapp/bin/bwa-0.7.12/bwa mem /bioapp/Data/155pathogens.fa $out/filter.fq >filter.bwa.sam\n"; system "perl /bioapp/bin/bwa_stat-v2.pl filter.bwa.sam /bioapp/Data/16S-complete-clean.taxon /bioapp/Data/pathogen.group filter.bwa.sam.stat\n"; print "Step 4: Report 报告生成\n"; system "cat filter.stat.genus filter.bwa.sam.stat.species >Report.txt\n";