大家好!今天我们来分享一篇发表在《Communications Biology》上的研究。做过宏基因组学分析的同学都知道,从样本处理到数据分析,每一步的选择都可能影响最终的微生物群落解读。实验流程和生信流程的标准化一直是领域内的热点和难点。这篇文章堪称“教科书式”的评估,系统性地比较了从DNA提取、文库构建、测序平台到生物信息学分析的全流程,覆盖了大家常用的Illumina短读长、PacBio和ONT长读长平台。更亮眼的是,作者还开发了一款名为“minitax”的生信小工具,旨在提供跨平台、跨方法的一致性物种注释结果。如果你正纠结于实验方案的选择,或者在为不同来源的数据如何整合分析而头疼,那么这篇研究绝对值得一读。它不仅给出了详尽的性能评估,还为不同需求(如成本、精度)的研究者提供了切实可行的方案建议。

摘要
背景:测序技术和生物信息学的飞速发展,让复杂的微生物生态系统宏基因组研究成为可能。然而,研究结果的可靠性高度依赖于一致的实验和生信分析流程。目前的方法优化往往只关注特定步骤,这使得我们很难全面理解每个环节对微生物种群分析的影响,也给跨研究的数据比较带来了挑战。
结果:这项研究使用了一个犬粪便样本、两种人工合成的微生物群落标准品以及多种来源的测序数据,对DNA提取方法、文库构建策略、测序平台和计算分析流程进行了全面评估。这是迄今为止对宏基因组学方法最全面的评估之一。研究团队开发了一款名为minitax的软件工具,它能够在不同的平台和方法学组合下提供一致的物种分类结果。研究发现,Zymo Research的Quick-DNA HMW MagBead试剂盒、Illumina DNA Prep文库构建方法与minitax生物信息学工具的组合,在获得高质量微生物多样性分析结果方面表现最为出色。
结论:然而,没有一种方法组合是放之四海而皆准的“最优解”,其效果会因样本类型而异。因此,为了在微生物系统研究中获得可靠的结果,采用多种方法进行验证或比较至关重要。
方法
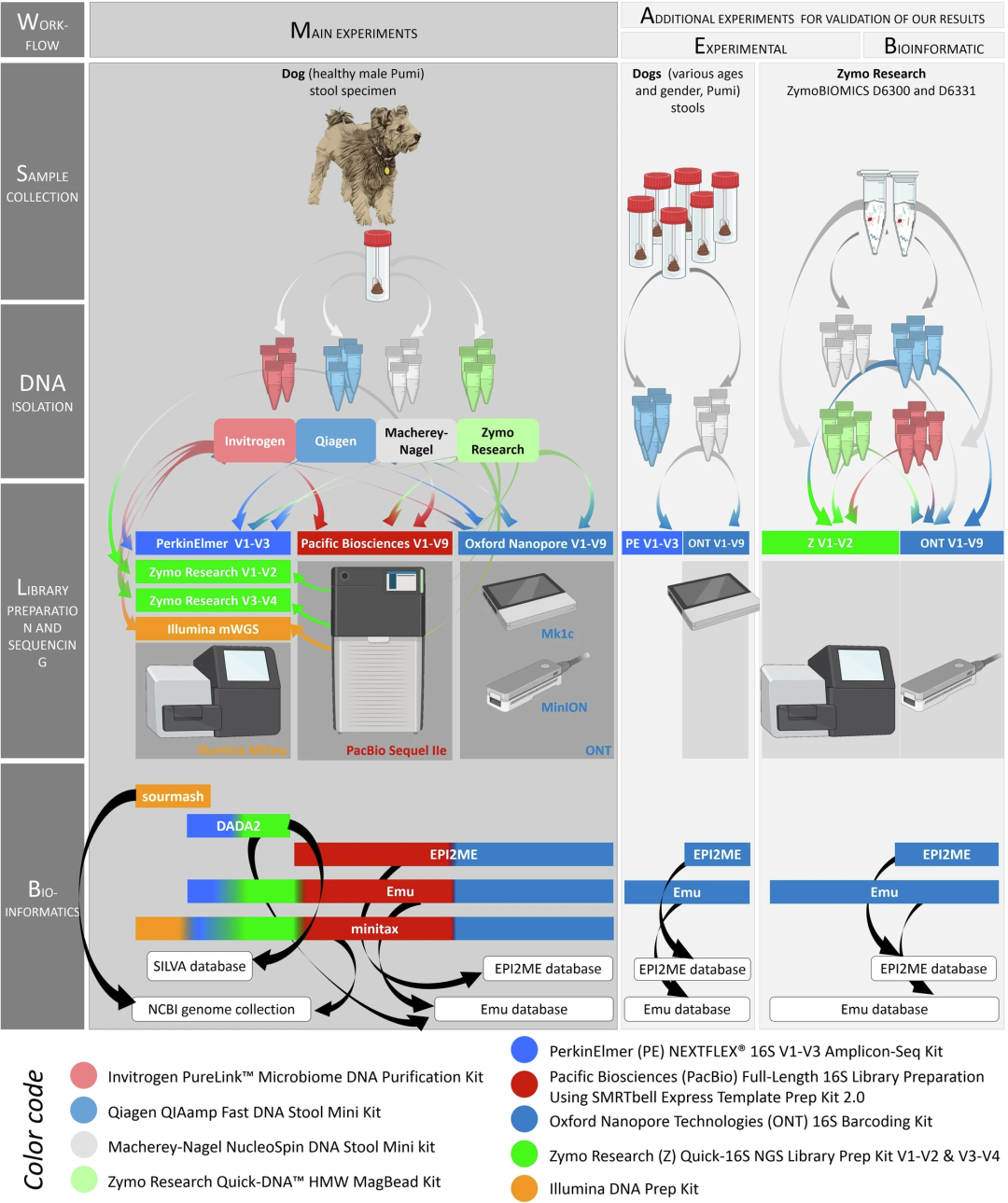
研究人员设计了一套精密的实验与分析流程来确保评估的全面性。他们选用了真实的犬粪便样本,并辅以两种商业化的微生物群落标准品(Zymo MCS,包含8个菌种;Zymo GMS,包含19个菌种)作为对照。
在DNA提取环节,他们对比了四种主流的商业试剂盒:QIAGEN的QIAamp Fast DNA Stool Mini Kit、Invitrogen的PureLink™ Microbiome Kit、Macherey-Nagel的NucleoSpin Stool Mini以及Zymo的Quick-DNA HMW MagBead Kit。每种试剂盒都设置了4个技术重复,以评估其稳定性和产量。
文库构建则涵盖了市面上多种主流方案,包括针对Illumina平台的mWGS(全基因组鸟枪法)、PerkinElmer的V1-V3区扩增子,以及Zymo的V1-V2和V3-V4区扩增子。同时,也兼顾了长读长平台,构建了针对ONT的V1-V9区16S rRNA基因扩增子文库和PacBio的SMRTbell V1-V9区16S文库。
测序在三大主流平台上进行:Illumina MiSeq、ONT MinION和PacBio Sequel IIe,确保了对短读长和长读长技术的全面覆盖。
生物信息学分析是本次评估的重中之重。对于Illumina 16S扩增子数据,他们使用了经典的DADA2流程(分别基于SILVA和Emu数据库进行物种注释)。对于WGS数据,则使用了sourmash。对于ONT和PacBio的长读长16S数据,则采用了Emu和EPI2ME流程。为了实现跨平台的统一比较,作者开发了核心工具minitax。该工具基于minimap2进行序列比对,通过评估MAPQ和CIGAR值来筛选高质量的比对结果,并提供了多种策略(如BestAln, RandAln, LCA)来确定最终的物种分类,最终输出可供下游phyloseq包直接使用的对象。下游的统计分析非常详尽,包括α和β多样性计算、PERMANOVA、PCoA、差异丰度分析(DESeq2)等,系统地评估了不同方法组合的性能差异。
主要结果
DNA提取方法的比较
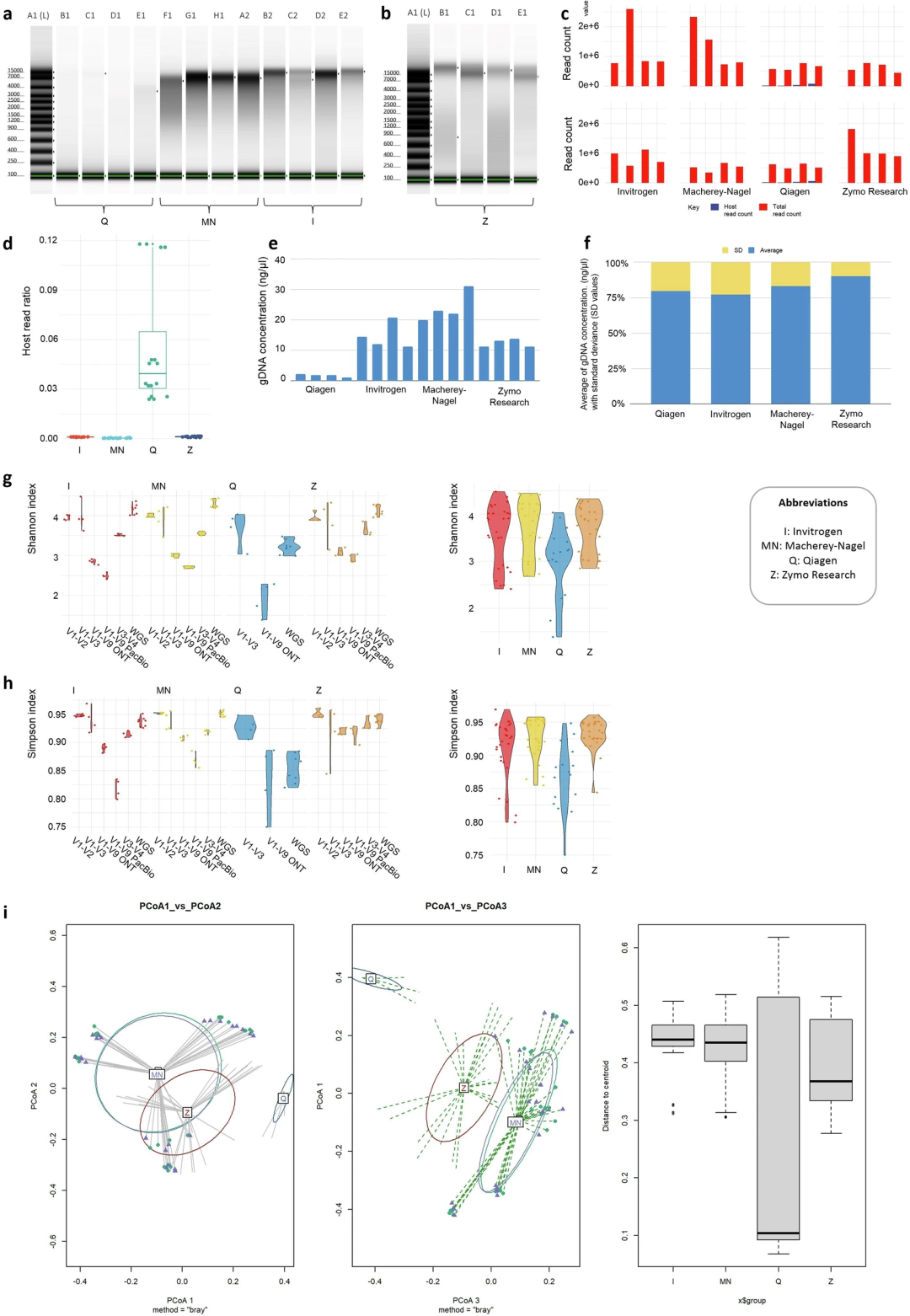 研究结果揭示了实验和分析流程中每一步的深远影响。在DNA提取阶段,Zymo试剂盒在DNA产量、片段长度和重复性方面综合表现最佳。相比之下,QIAGEN试剂盒的产量最低,且宿主DNA污染较高,导致其在α多样性评估中显著降低了物种的丰富度和均匀度。从β多样性来看,DNA提取方法本身解释了群落结构差异的28.2%,其中QIAGEN提取的样本在PCoA排序图中明显偏离其他三组。另外就是不同方法对革兰氏阳性菌和革兰氏阴性菌比例偏好性。
研究结果揭示了实验和分析流程中每一步的深远影响。在DNA提取阶段,Zymo试剂盒在DNA产量、片段长度和重复性方面综合表现最佳。相比之下,QIAGEN试剂盒的产量最低,且宿主DNA污染较高,导致其在α多样性评估中显著降低了物种的丰富度和均匀度。从β多样性来看,DNA提取方法本身解释了群落结构差异的28.2%,其中QIAGEN提取的样本在PCoA排序图中明显偏离其他三组。另外就是不同方法对革兰氏阳性菌和革兰氏阴性菌比例偏好性。
文库构建和测序方法的评估
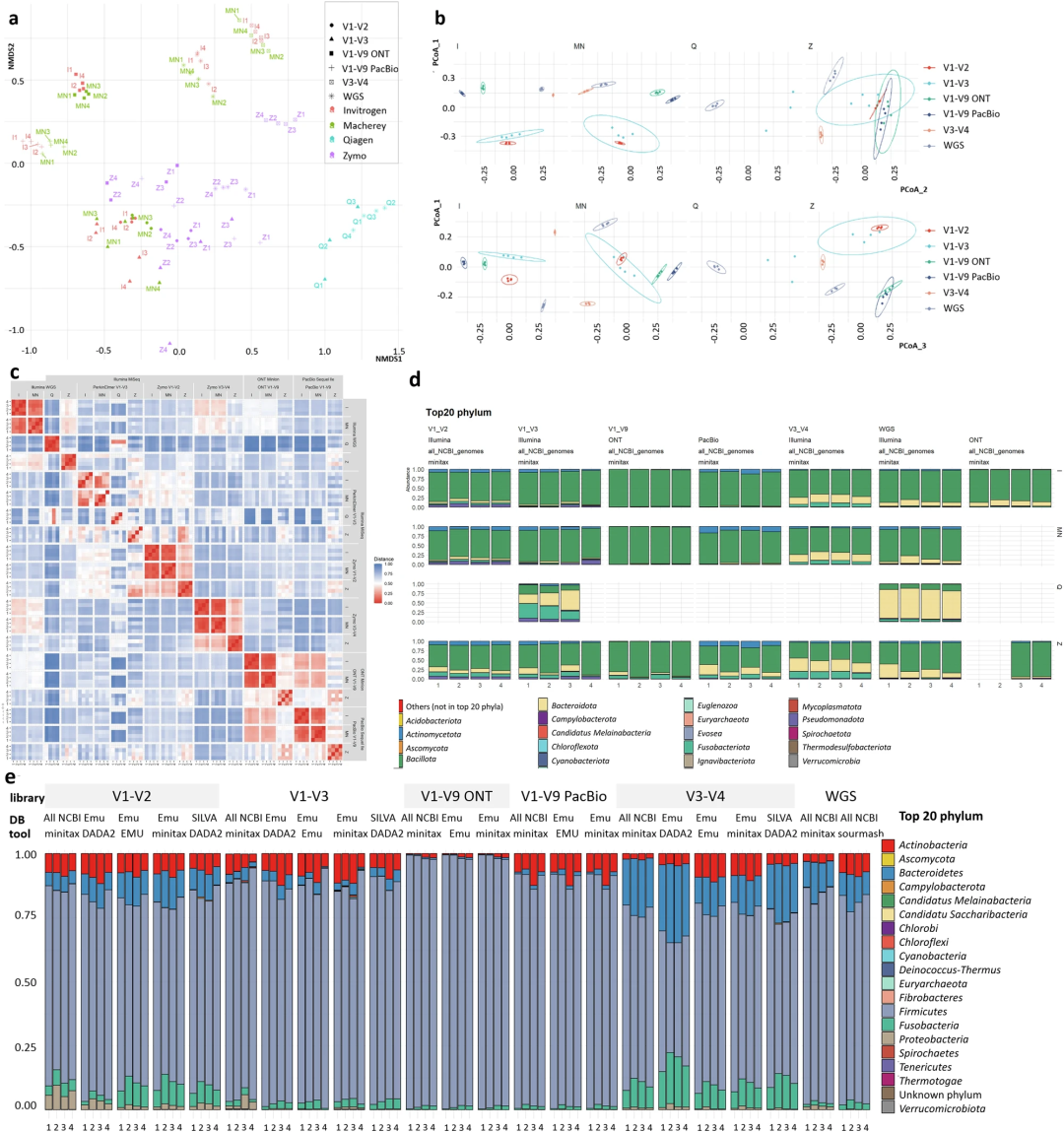
文库构建和测序平台的影响甚至超过了DNA提取,解释了高达58.8%的群落结构变异。特别是16S rRNA基因的不同扩增区域(如V3-V4 vs. V1-V2/V1-V3)导致了最显著的群落分离。
在合成菌群落的评估中,不同方法表现各异。对于物种较少的MCS标准品,Invitrogen的提取方法最为接近理论组成;而对于更复杂的GMS肠道标准品,Zymo方法的偏差最小。这表明,标准品的评估结果并不能简单地推广到所有样本类型,方法的选择需要因“材”而异。
生信分析的比较评估
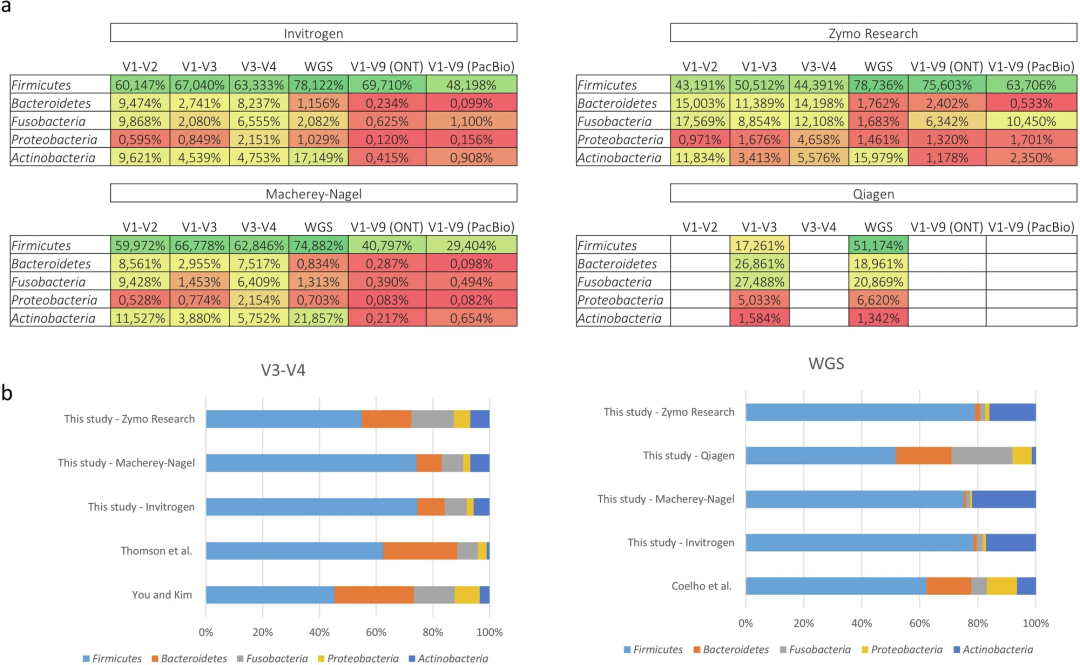
在生物信息学分析层面,分析流程和所用数据库的差异是造成结果不同的最主要因素,解释了59.4%的群落变异。例如,使用DADA2结合SILVA数据库的结果与基于Emu或minitax的注释结果差异显著。作者开发的新工具minitax在跨平台数据整合分析上展现了其价值。无论是在ONT、Illumina还是PacBio的数据上,minitax都表现出稳健和一致的性能,尤其适合作为不同来源数据进行统一比较分析的“中间件”。例如,在CAMISIM模拟数据集上,minitax处理后的不同平台数据在门水平(Phylum)的相关性(r²)高达0.96。
结论
这项研究为宏基因组学研究者提供了非常实用的指导。总的来说,Zymo的Quick-DNA HMW MagBead Kit被推荐为综合性能最佳的DNA提取试剂盒。在文库构建方面,需要根据成本和精度需求进行权衡:ONT V1-V9扩增子测序成本效益高但质量稍逊,而Illumina WGS和PacBio 16S则能提供极高精度但成本也更高。
在生物信息学分析方面,对于全长或多区域的16S扩增子数据,推荐使用Emu;对于WGS数据,sourmash是优选;而当需要整合比较来自不同平台或方法的数据时,minitax凭借其一致性成为最有力的工具。作者最后给出了贴心的组合建议:若成本敏感,可选择Zymo提取+ONT V1-V9测序;若追求平衡,可采用Invitrogen提取+Illumina V3-V4测序;若要求最高精度,则推荐MN提取+Illumina DNA Prep WGS的方案。当然,这只是作者的建议,小编相信各位有自己的见解。
数据集与代码
研究的所有测序数据都已提交至ENA数据库,项目号为PRJEB59610。所有用于生成图表的分析数据和phyloseq对象都可以在GitHub上找到( https://github.com/Balays/Microbiome-Method-Comparison)。核心的生信工具minitax也已开源,地址为: https://github.com/Balays/minitax。
参考文献
-
Gulyás, G., Kakuk, B., Dörmő, Á. et al. Cross-comparison of gut metagenomic profiling strategies. Commun Biol 7, 1445 (2024). https://doi.org/10.1038/s42003-024-07158-6
本篇文章来源于微信公众号:微因