今天查看gwas-catalog网站,发现即将推出GWAS 汇总统计数据的新格式标准GWAS-SSF[1],困扰大家多年的格式不统一问题有望解决啦?暂时还未发表,还在预印本服务器,让我们先一睹为快!
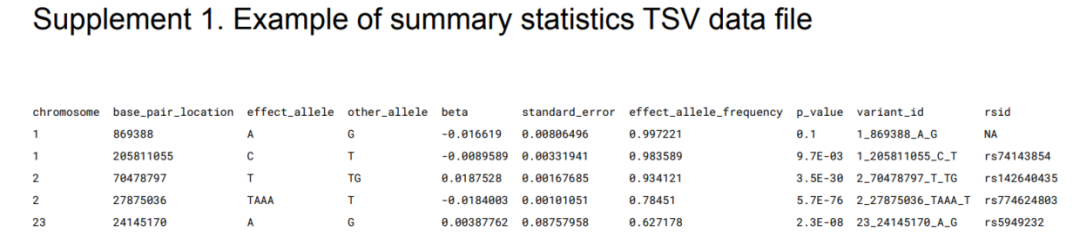 全基因组关联研究(GWAS)的汇总统计数据代表了巨大的研究潜力。该领域研究人员面临的挑战是,由于缺乏数据内容和文件格式的标准,汇总统计数据的访问和共享。出于这个原因,GWAS目录在2021年与汇总统计利益攸关方举行了一系列会议,以指导标准格式的开发。利益攸关者的主要要求是,一个包含关键数据要素的标准能够支持广泛的数据分析,需要较低的生物信息学技能来访问和生成文件,拥有易于获取的元数据,以及明确和可互操作的数据。在这里,我们定义了 GWAS-SSF 格式第一版的规范,该格式的开发是为了满足与社区讨论的要求。GWAS-SSF 由一个制表符分隔的数据文件组成,该文件具有明确定义的字段和随附的元数据文件。
全基因组关联研究(GWAS)的汇总统计数据代表了巨大的研究潜力。该领域研究人员面临的挑战是,由于缺乏数据内容和文件格式的标准,汇总统计数据的访问和共享。出于这个原因,GWAS目录在2021年与汇总统计利益攸关方举行了一系列会议,以指导标准格式的开发。利益攸关者的主要要求是,一个包含关键数据要素的标准能够支持广泛的数据分析,需要较低的生物信息学技能来访问和生成文件,拥有易于获取的元数据,以及明确和可互操作的数据。在这里,我们定义了 GWAS-SSF 格式第一版的规范,该格式的开发是为了满足与社区讨论的要求。GWAS-SSF 由一个制表符分隔的数据文件组成,该文件具有明确定义的字段和随附的元数据文件。
汇总统计表内容
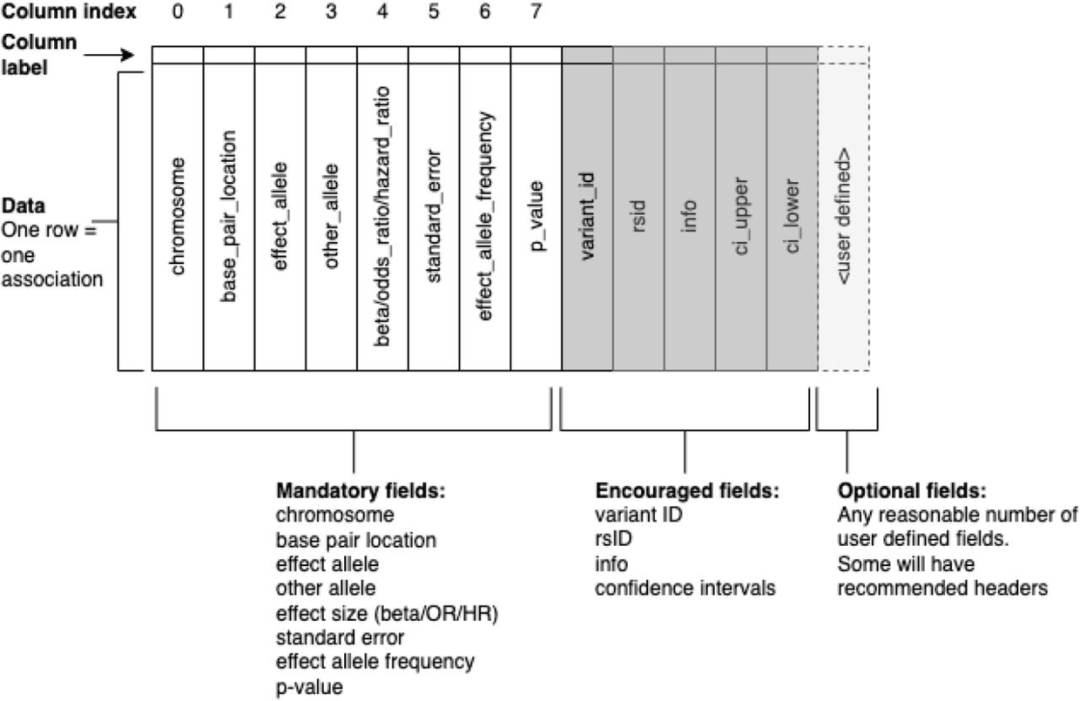 汇总统计表中的四个字段,结合元数据文件中提供的参考基因组,明确定义了遗传变异。这些字段是染色体(染色体)、染色体上的基因组位置 (base_pair_location)、效应等位基因 (effect_allele) 和非效应等位基因 (other_allele)。染色体值是从 1 到 25 的整数,染色体 X 映射到 23,染色体 Y 映射到 24,线粒体映射到 25。基因组位置是一个整数值,代表变异在参考基因组中的第一个位置,使用基于1的索引。以最大限度地提高与变异调用格式(VCF)的互操作性(Danecek等人,2011[2])。effect_allele字段捕获与效应相关的等位基因,而other_allele场报告非效应等位基因。两个等位基因字段都将包含等位基因字符串,包括变体为插入和删除的情况。这四个字段(染色体、base_pair_location、effect_allele、other_allele)连接起来以填充variant_id字段,rsID 可以存储在 rsid 字段中,但这两个字段都是可选的。
汇总统计表中的四个字段,结合元数据文件中提供的参考基因组,明确定义了遗传变异。这些字段是染色体(染色体)、染色体上的基因组位置 (base_pair_location)、效应等位基因 (effect_allele) 和非效应等位基因 (other_allele)。染色体值是从 1 到 25 的整数,染色体 X 映射到 23,染色体 Y 映射到 24,线粒体映射到 25。基因组位置是一个整数值,代表变异在参考基因组中的第一个位置,使用基于1的索引。以最大限度地提高与变异调用格式(VCF)的互操作性(Danecek等人,2011[2])。effect_allele字段捕获与效应相关的等位基因,而other_allele场报告非效应等位基因。两个等位基因字段都将包含等位基因字符串,包括变体为插入和删除的情况。这四个字段(染色体、base_pair_location、effect_allele、other_allele)连接起来以填充variant_id字段,rsID 可以存储在 rsid 字段中,但这两个字段都是可选的。
汇总统计数据元数据

汇总统计数据文件附带一个附加文件,其中包含描述汇总统计数据的元数据,例如汇总统计数据文件的名称和md5sum和GWAS元数据本身,包括样本和实验元数据,从而确保数据的可重用性。元数据文件字段将来可以根据需要展开,并且与汇总统计数据文件一样,可以根据需要包含其他列。示例元数据字段包括所调查性状以及样本大小和血统的描述。可以使用额外的字段血统方法来指示血统描述符是自我报告的还是遗传定义的(鼓励)。我们建议根据Morales等人,2018[3]年描述的标准化框架指南报告血统。应尽一切努力明确注意样品是否混合以及导致外加剂的祖先背景。性状描述是自由文本,应包括对所研究性状的清晰描述,包括研究人群的任何相关背景特征,例如“哮喘患者的肺癌”。特征本体术语可以存储在元数据 ontologyMapping 字段中。元数据文件采用 YAML 格式,这是“适用于所有编程语言的人性化数据序列化语言”[4]。
是不是考虑用起来呢?阅读原文是预印本文章地址。
参考资料
GWAS-SSF: https://www.biorxiv.org/content/10.1101/2022.07.15.500230v1
[2]Danecek等人,2011: https://www.biorxiv.org/content/10.1101/2022.07.15.500230v1.full#ref-3
[3]Morales等人,2018: https://www.biorxiv.org/content/10.1101/2022.07.15.500230v1.full#ref-9
[4]http: https://yaml.org/
本篇文章来源于微信公众号:微因