最近涉猎了些扩增子甲基化的测序的内容,发现单就分析方面,就有不少的分析流程呢,一一列举一下!
1、amplikyzer2
这是一个从焦磷酸测序走来的流程,第 2 版增加了对 illumina MiSeq 数据的支持,不知道效果,有待测试,其可以完成从数据拆分到最后的结果展示,是一个比较完善的流程,甚至还有图形界面。
usage: amplikyzer2 [-h] [--path PATH] [--conf FILE [FILE ...]] [--version] {analyzefastq,statistics,methylation,align,analyzesff,printsff} ...
amplikyzer2: an amplicon analyzer
positional arguments:
{analyzefastq,statistics,methylation,align,analyzesff,printsff}
analyzefastq analyze .fastq files (identify MID, tag, primer, ROI for each read)
statistics show statistics for an analyzed dataset
methylation do a methylation analysis of a given locus and MID
align output a multiple alignment of all reads of a locus for a given MID
analyzesff analyze .sff files (identify key, MID, tag, primer, ROI for each read)
printsff print reads of an .sff file
optional arguments:
-h, --help show this help message and exit
--path PATH, -p PATH project path (directory) containing read files
--conf FILE [FILE ...]
names of configuration files with MIDS, TAGS, LOCI, LABELS
--version show program's version number and exit
In development. Use at your own Risk!
比较好奇它的步骤是不是可以路过,比如这个数据拆分,看说明是不可以跳过的,不过有点不理解意思是啥,一起研究下呀!
“
MiSeq-FASTQ specific features: Samples can be de-multiplexed from barcodes in filenames.
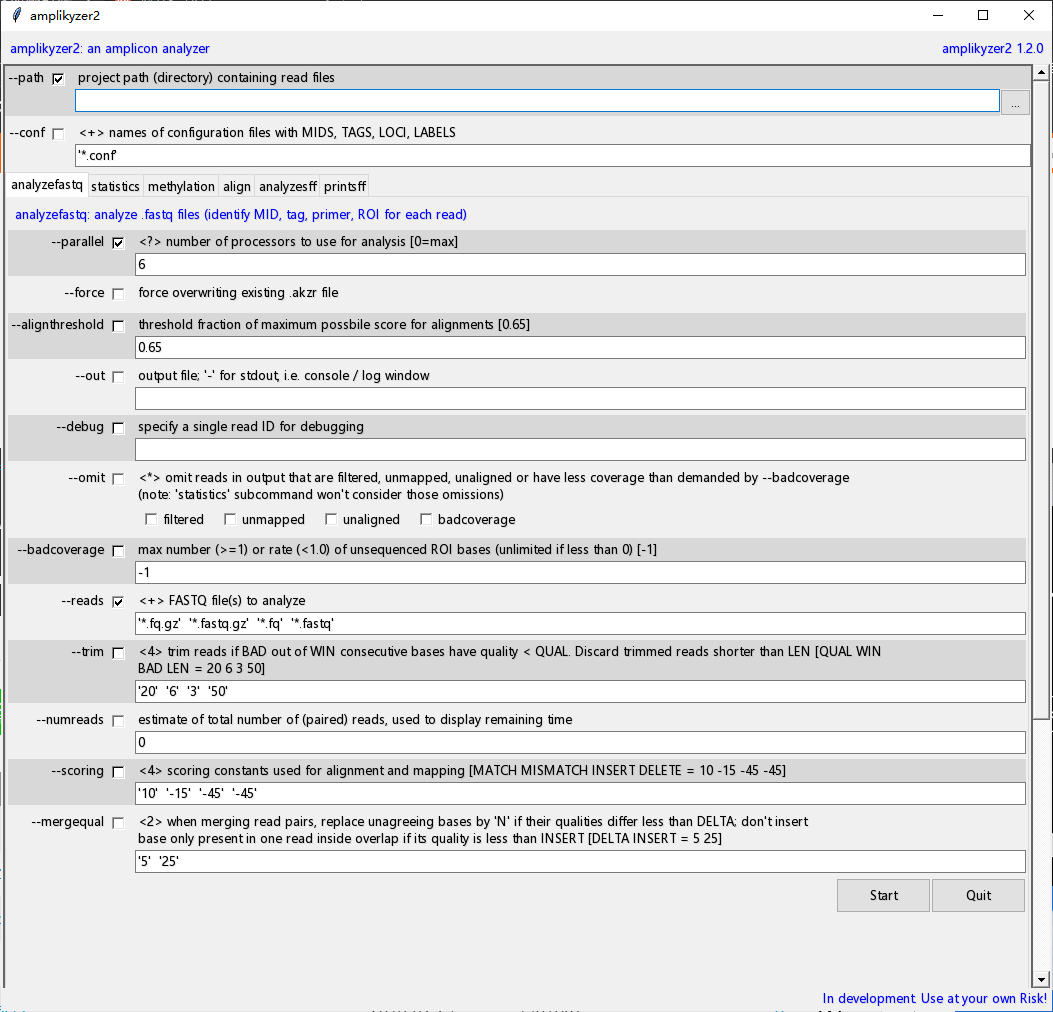 值得一提的是,作者号称它的图形界面可以兼容其他 python 程序,可以试试呀!附上官方镜像:svenrahmann / amplikyzer / wiki / Home — Bitbucket[1]
值得一提的是,作者号称它的图形界面可以兼容其他 python 程序,可以试试呀!附上官方镜像:svenrahmann / amplikyzer / wiki / Home — Bitbucket[1]
2、AmpliMethProfiler[2]
这个,从名字也可以看出它是做啥的,同样是 python 写的,需要本地安装 BLAST 和 (可选) QIIME 工具。处理流程如下,同样也能拆数据,有些借鉴了扩增子测序数据处理的方法的感觉。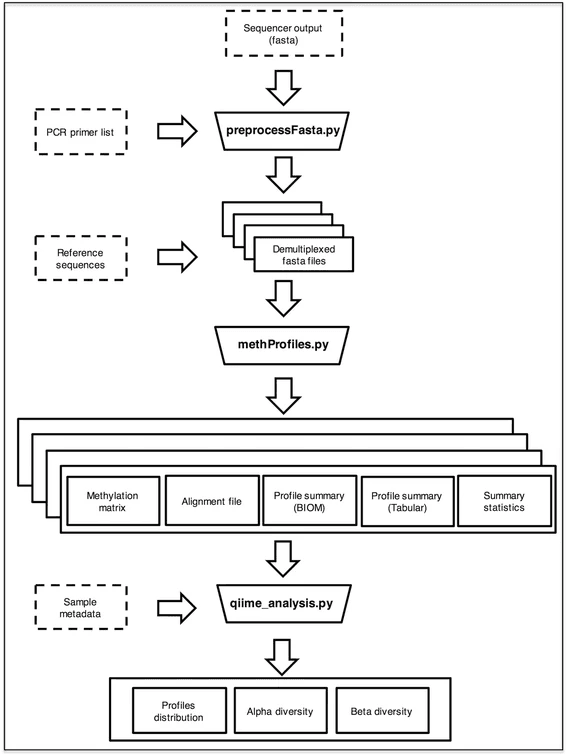 下面是文章的分析结果,看起来还不错!
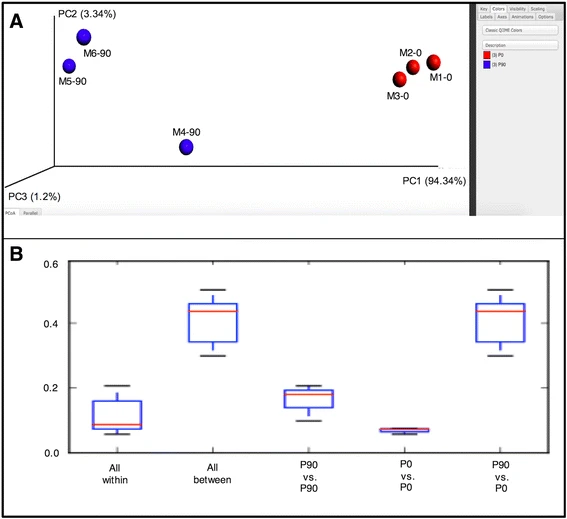
下面是文章的分析结果,看起来还不错!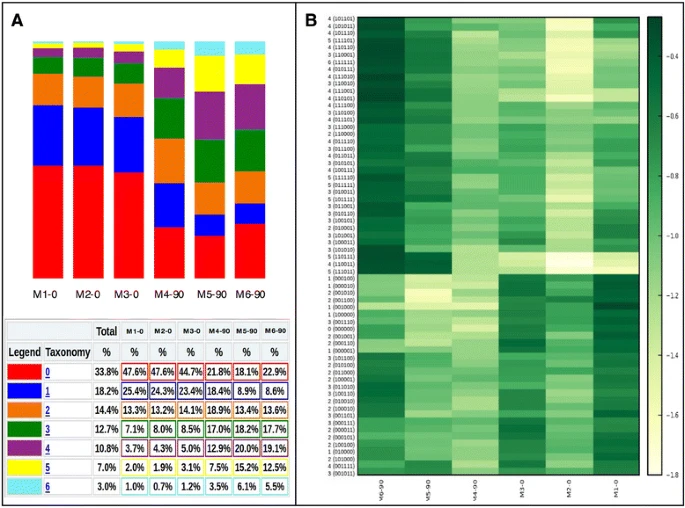
 作者还列了个文章发表时已有的软件对比,看起来还真不少,不过主流的,好用了,估计也就一两个啦!
作者还列了个文章发表时已有的软件对比,看起来还真不少,不过主流的,好用了,估计也就一两个啦!
3、EPIC TABSAT (ait.ac.at)[3]
这个新版本直接是个网页工具,像许多公司搞的云平台了,比较赞!旧版本是开源并可以使用命令行的,二者的算法应该区别不大,区别估计在于易用性方面。下图是数据处理的一个流程,可以看到由质控、引物比对、reads 处理。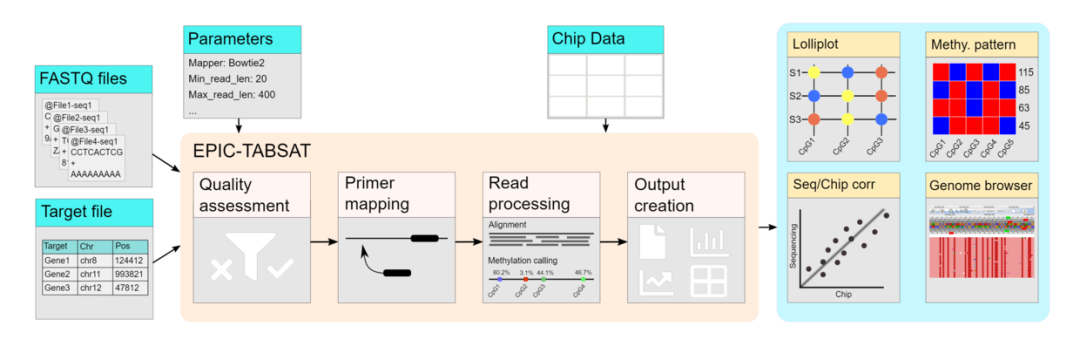 下图是网页全貌。
下图是网页全貌。
4、IMPLICON – bisulfite amplicon data for imprinted loci
 由上图的机构编写的,应该是个大牛机构啦!同样是一个比较完整的流程,因为针对超深度测序,还支持 umi 标签,不确定灵活性如何。
由上图的机构编写的,应该是个大牛机构啦!同样是一个比较完整的流程,因为针对超深度测序,还支持 umi 标签,不确定灵活性如何。 示例分析结果如下图:
示例分析结果如下图: 地址在这:FelixKrueger/IMPLICON: A processing guide for IMPLICON data (bisulfite amplicon data for imprinted loci) (github.com)[4]
地址在这:FelixKrueger/IMPLICON: A processing guide for IMPLICON data (bisulfite amplicon data for imprinted loci) (github.com)[4]
5、其他流程
1)rose-driscoll/methylation-analysis: Data processing pipelines and statistical analysis for a bisulfite amplicon sequencing methylation analysis study (Driscoll et al 2019 [submitted]) (github.com)[5]2)okon/MethAmplicons: Analysis pipeline of NGS amplicon data for methylation analysis (github.com)[6]根据李金明主编的《高通量测序技术》中的描述,甲基化测序的比对与正常测序有所不同,因为经过了重亚硫酸盐处理,不同的软件原理有所区别,贴这一起学习下啦!


欢迎加微信交流:

欢迎加微信交流:
参考资料
svenrahmann / amplikyzer / wiki / Home — Bitbucket: https://bitbucket.org/svenrahmann/amplikyzer/wiki/Home
[2]
AmpliMethProfiler: https://sourceforge.net/projects/amplimethprofiler/
[3]
EPIC TABSAT (ait.ac.at): https://tabsat.ait.ac.at/
[4]
FelixKrueger/IMPLICON: A processing guide for IMPLICON data (bisulfite amplicon data for imprinted loci) (github.com): https://github.com/FelixKrueger/IMPLICON
[5]
rose-driscoll/methylation-analysis: Data processing pipelines and statistical analysis for a bisulfite amplicon sequencing methylation analysis study (Driscoll et al 2019 [submitted]) (github.com): https://github.com/rose-driscoll/methylation-analysis
[6]
okon/MethAmplicons: Analysis pipeline of NGS amplicon data for methylation analysis (github.com): https://github.com/okon/MethAmplicons
本篇文章来源于微信公众号: 微因