前面做的许多处理基本上自己拼凑来的,下面再看下完整解决方案。researchgate网站上有人说qiime1版本有这个双向数据配对不拼接的选项?这个没找到。主要发现了有两个方案,一个是有篇文章提出了一个流程Hybrid-denovo,还有一篇peer review的文章,几个人评议还有一个人不同意,anyway,都看下。
1.Hybrid-denovo流程处理nonoverlapping 扩增子[2]
这里其实主要指的是为了提高精度测几个V区,比如 V3-V5这种,找到了一篇梅奥医学的同行发表在华大Giga Science上的文章,看名字作者中有多个华人,也学习下。其实这篇文章主要是要充分利用双向测序数据和质控去除的不配对单向数据的,也可以用于单纯地双向nonoverlapping数据。由于OTU聚类已经不是未来研究的主流,所以,这个方法经典,但以后可能要用ASV。

摘要
背景
Illumina-16S双向测序数据相比单向测序提供了更好的物种分辨率,这是因为有效测序长度的增加。但是,反向测序数据R2的质量相比正向数据R1会有迅速地下降,很大比例地反向测序数据会在质控中被丢弃,导致clean reads成为Paired-end reads和Single-end reads的混合。一个典型的16S数据分析流程一般是只处理Paired-end reads或者Single-end reads数据。这样,定量的准确性和统计上的说服力会由于这些数据的损失而减小。结果是,罕见的物种Paired-end reads数据可能检测不到,而Single-end reads数据会导致低的物种分辨率。
结果
为了充分利用Paired-end reads高分辨率和Single-end reads高覆盖率的优势,提出了一个OTU-picking的流程hybrid-denovo,可以处理Paired-end reads和Single-end reads的混合。使用高质量的Paired-end reads作为金标准,结果显示hybrid-denovo和金标准的一致性最高,在物种多样性和丰度上,比单独Paired-end reads或者Single-end reads数据表现更好。应用于类风湿性关节炎(RA)数据集, hybrid-denovo流程也检测了更多的微生物多样性,更多的RA-相关菌。
结论
hybrid-denovo充分利用了Paired-end reads和Single-end reads,推荐用于分析16S rRNA基因双向测序数据。
简介
16S数据分析中,OTU(操作分类单元)聚类仍然是一个主要部分,有de novo和基于参考序列的两种,前者基于序列相似度,不需要参考序列,产生的OTU能更好地和数据比对,然而,需要对同一基因区域进行比较。后者可以克服这个,可是依赖于一个建好的OTU代表序列数据库,可能对一些特定数据集不适合。
2013-2015年期间,梅奥医学的数据集中,只有24%的R2数据通过了质控,而R1有83%。我们一般是只用Paired-end 一小部分数据和测序深度更高的R1数据进行分析。为了充分利用Paired-end reads和Single-end reads的优势,最大化地检出罕见物种,我们提出了hybrid-denovo。
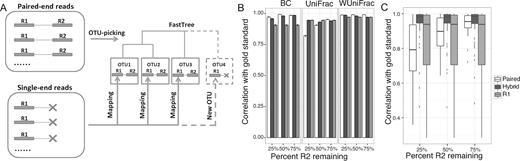
A图是hybrid-denovo的流程图,B图是使用金标准加三种不同比例的高质量R2数据获得的beta多样性矩阵Mantel相关性检验(unweighted UniFrac, weighted UniFrac, and Bray-Curtis),误差条表示基于100引导的估计的标准误差。C图是56个流行属相对丰度的相关盒形图
方法
1.仅使用paired-end reads,构建OTU骨架,余下的single-end reads (R1)比对到这个OTU骨架上,如果没比对上,建立新的OTU,和IM-TORNADO方法使用相同的质控和OTU聚类过程。具体就是,具体来说,使用Trimmomatic进行质量过滤,对5’和3’末端的读数严格截断PHRED得分Q3,以移动平均得分Q15修剪3’末端,窗口大小为4个碱基 ,并删除所有少于原始读取长度75%的剩余读取。去除任何不确定的碱基,统一两个reads的长度,然后进行连接,按照簇大小排序。
2.使用UPARSE算法进行de novo聚类,基于RDP Gold数据库参考序列进行UCHIME去嵌合,得到一个高质量的OTU代表序列。
3.使用USEARCH把单向R1数据比对到OTU序列的R1端(如果有多个具有相同得分的匹配,则默认情况下将选择最丰富的匹配),其余的未比对上的R1通过UPARSE算法聚集到新的OTU中,并添加到由配对末端读取生成的OTU列表中。因此,OTU是长短序列两者的混合,然后,我们使用在核糖体数据库项目(RDP)的数据库上训练的结构比对算法对齐所有OTU表。未比对上的OTU序列被删除,因为认为它们代表非细菌
4.FastTree构建系统发育进化树,FastTree对末端gap的影响很小,这在处理单端读取和成对读取的混合时非常有利。
5.最后,将R1和R2用歧义核苷酸(一串Ns)连接在一起,然后由RDP分类器Greengenes分配分类。
6.未归类为细菌的和singleton OTU被认为是污染物,因此被删除。注意,此步骤可能丢失了数据库中未表示的多样性,这是准确性和完整性之间的权衡。
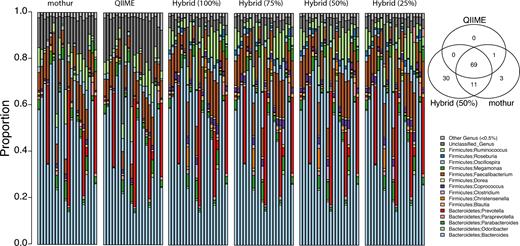
在属水平比较mothur, QIIME和hybrid-denovo, Hybrid-denovo在具有不同百分比的高质量R2读取(100%,75%,50%和25%)的数据集上运行。 每列代表在所有重复样本中平均值的个体的微生物群谱。 维恩图中显示了3个流程之间检测到的属的重叠。
为了验证我们的方法,我们基于在Mayo核心实验室(V3-V5 16S扩增子,694nt,357F / 926R引物)上测序的837个高覆盖率人类粪便样品,创建了具有高质量配对末端读数的金标准数据集 )。这些粪便样本使用6种不同方法(无添加剂,RNAlater,70%乙醇,EDTA,干拭子和粪便潜血试验[FOBT])从20位受试者中收集。立即将样品冷冻或在室温下保存4天,以研究微生物群的稳定性。 每个条件都有2-3个技术重复,以评估可重复性。我们运行Trimmomatic进行质量控制,并将R1降低至250 bp,将R2降低至200 bp,以确保较高的碱基质量,从而实现了非重叠的配对末端reads。 对于每个样本,我们获得了8000个高质量的配对末端读数。然后,我们使用IM-TORNADO基于这些配对末端读段执行OTU拣选和分类法分配。 这些最终的OTU及其相关的分类法构成了黄金标准数据集。 然后,我们将剩余的R2读数的25%,50%和75%划分为黄金标准。这3个子数据集代表了实践中遇到的R2质量的不同级别。我们比较了基于单端R1或使用子数据集的双端读取的从头方法与Hybrid-denovo方法。通过计算Spearman与金标准在微生物β多样性(未加权和加权的UniFrac和Bray-Curtis距离)和属水平相对丰度方面的相关性来评估性能。

我们还将我们的管道与QIIME和mothur(分别为1.8.0版和1.39.3版)对金标准数据进行了比较。由于QIIME和mothur目前不支持基于非重叠读取的从头OTU聚类,因此我们在R1读取中运行QIIME和mothur。选择的参数设置与Hybrid-denovo的设置相当。当我们使用Trimmomatic创建高质量的读取文件时,通过不应用其他读取QC过滤器,我们减少了管道之间性能的潜在差异。使用经过Greengenes v13.5训练的RDP分类器对所有管道的读取进行分类。滤除singleton和非细菌OTU(基于分类法)。补充说明1中记录了除了用于重现结果的命令外,这三个管道之间的主要区别。


我们通过研究(1)在属水平上检测到的属的数目和未分类读物的百分比,(2)使用Bray-Curtis(BC)矩阵的Mantel相关性,以及(3)这些的类内相关系数(ICC)来评估性能 在超过90%的样本中观察到了核心OTU和属。ICC是技术复制之间相关性的一种度量。较高的值表示较小的测量误差。 使用R包 ICC计算ICC。
最后,我们在风湿性关节炎(RA)患者粪便微生物组研究的数据集上论证了该方法的性能,该数据集由40位RA患者和49位对照(V3-V5 16S扩增子,694 nt)组成。我们将DESeq2应用于分类单元计数数据以进行差异丰度分析,并比较了RA相关的OTU和通过不同方法回收的属。
结果
三种方法中,微生物β多样性与金标准的相关性通常都很高(图1B)但是,当使用BC距离时,基于单端R1的方法往往具有较低的相关性(单端R1方法对于R2的数量不变)。另一方面,当仅保留25%的R2时,成对末端方法与未加权UniFrac的相关性要低得多。这是由于以下事实:未加权的UniFrac主要决定于群落成员,这主要是由稀有分类单元贡献的,并且由于读取丢失,成对末端方法不再能够检测到许多稀有分类单元。相反,Hybrid-denovo非常出色,并且在两种多样性指标中与黄金标准的相关性最佳或接近最佳。对于加权UniFrac距离,这三种方法的相关性都相似,因为加权UniFrac受占比例大的分类单元的影响最大,并且所有方法都很好地量化了这些显性分类单元(图1B)。
接下来,我们研究了所提出方法的分类分析性能。基于56个属的出现率大于10%,hybrid-denovo在所有考虑的场景中均与金标准具有更高的相关性,并且其性能对剩余的R2s百分比不是很敏感(图1C)。相反,成对端方法的性能在很大程度上取决于R2质量,而当R2质量低时,其相关性要低得多。单端R1方法对于预期的R2数量是不变的,并且仅在R2质量较低时才比配对端方法更好,补充图3是属水平的相关性。对于单端方法,2个属与金标准显示0相关性,因为它们的所有R1由于其较短的长度而被重新分类到科级别(Lachnobacterium映射为Ruminococcaceae,Erwinia映射为Enterobacteriaceae),这表明系统发育分辨率提高了 使用配对末端读取。对于配对末端方法,具有低丰度的属表现出较低的相关性,表明由于配对末端读数的丢失而降低了定量准确性。

mothur,QIIME和hybrid-denovo对核心属(A)和OTU(B)的类内相关系数(ICC)的比较。 ICC是根据6种不同粪便收集方法的技术重复计算得出的。 Hybrid-denovo在具有不同百分比的高质量R2读取(100%,75%,50%和25%)的数据集上运行。
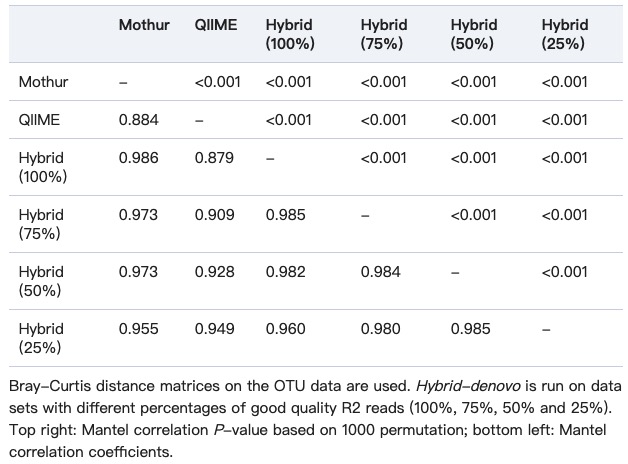
基于金标准数据集,我们还比较了mothur和QIIME(两种用于16S数据的主要管道)与hybrid-denovo。Mothur和QIIME分别花费了大约24和6个小时来完成金标准数据集(n = 837)的分析,而我们的流程大约需要1个小时。Mothur和QIIME分别总共生成了4599和2898个非单OTU,而hybrid-denovo在具有不同质量的R2读取百分比的数据集上分别生成了1094、1086、1079和1049个非单OTU(100%,75%,50 %和25%)。尽管我们的流程生成的OTU数量较少,但我们检测到的属数比mothur和QIIME多。例如,将hybrid-denovo应用于具有50%优质R2读数的数据集,共产生110属,而QIIME和mothur分别为70和84(图2,右上方,维恩图)。在针对Greengenes数据库的QIIME和mothur特定属(基于R1读物进行分类)的配对末端配对中使用BLAST,会将许多reads重新分配给其他属。这表明这些属可能由于读长短而被错误分类。尽管所有管道中20位受试者的菌群水平菌群谱均相似(图2),但hybrid-denovo的具有未知菌种(5%)的reads比例分别要比mothur和QIIME(14%和18)低得多 。综上所述,这些观察结果表明,由于使用更长的读段,杂交-denovo具有更高的分类学分辨率。有趣的是,所有管道都可以产生相似的样本间关系,这是根据基于Bray-Curtis距离矩阵的Mantel相关系数测得的(表1)。数据集技术复制的可用性使我们能够使用类内相关系数比较不同的管道。高ICC表示由生物信息学渠道引入的变异性较小。我们计算了核心OTU和属的不同粪便收集方法的ICC,这些ICC发生在90%以上的样本中。我们的管道通常比mothur和QIIME具有更高的ICC(技术复制之间的差异较小)(图3)。相反,在核心OTU和属上,mothur和QIIME的性能分别不佳。
表格1:QIIME,Mothur和Hybrid-denovo之间样本间距离的Mantel相关性。
我们还将我们的方法应用于来自RA研究的数据集[18],其中质量控制后约有40%的R2被丢弃(补充表1)。如预期的那样,hybrid-denovo产生OTU和属的数量最多(图4A),并且涵盖了成对端方法的所有属和单端R1方法的大多数属(图4C)。在这5个R1特异属中,将梭状芽孢杆菌梭菌科和梭状芽孢杆菌梭菌科02d0细菌重新分类为梭状芽胞杆菌梭菌科的梭状芽胞杆菌属,由于R1读长短导致错误分类。
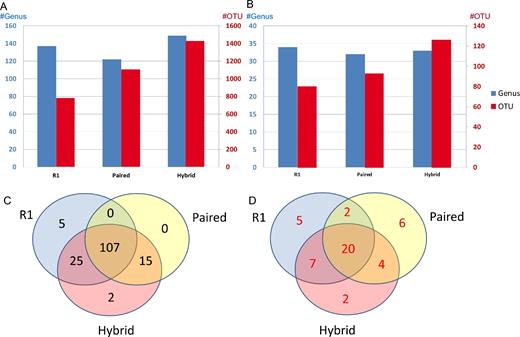
RA数据集上R1,配对和混合方法的比较。 A,检测到的OTU数量(红色)和属(蓝色)。 B,通过差异丰度分析(FDR≤0.01)得出的有效OTU数(红色)和属(蓝色)。 C,检测到的属的维恩图。 D,通过差异丰度分析得出的重要属的维恩图。
除了比较检测到的属外,我们还证明了在使用DESeq2进行差异丰度分析的背景下,我们流程的优势。
我们从测试中排除了少于10%样本中出现的OTU。分别使用混合Denovo,配对和R1方法测试了总共758、578和393个OTU。由于更高的读取计数和更高的系统发育分辨率,杂交denovo恢复了更多的差异OTU。在FDR调整后的P值为0.01的情况下,我们总共确定了126个重要的OTU,而对于双端和单端R1方法,分别为93和80个OTU。由于不同的方法具有自己的OTU定义,并且很难直接比较差分OTU,因此我们比较了已识别OTU的属身份。混合-denovo鉴定出的差异性OTU分为33属,而双端和单端R1方法分别为32和34(图4B)。这3种方法共有20个重要属(图4D),其中许多是以前的研究报告的。例如,拟杆菌中的对照样品中富集,而柯林斯菌属,埃格特氏菌,普雷沃特氏菌和梭状芽胞杆菌中的RA样品则富集。 即使所有方法的差异属的总数相似,我们的流程仍可确定其他两种方法中的一种均共享的最多属(n = 11),而配对末端和配对方法分别为6和9。 这表明杂交-denovo方法能够识别被双端R1方法或单端R1方法遗漏的差异属。此外,hybrid-denovo与配对末端(n = 6)和R1单末端(n = 5)的比较中,方法特定属的数目最少(n = 2)。由于缺乏其他方法的支持,特定于方法的属可能不太可靠。例如,R1方法发现Veillonella富含对照样品,这与先前的研究相矛盾。有趣的是,在Zhang等人的报道中,在两个杂种-denovo特异性属中,克雷伯氏菌在健康人群中富集。
讨论
我们提出了基于配对末端16S序列标签的从头OTU挑选的Hybrid-denovo。 通过仿真和实际数据示例,我们证明了在定量微生物多样性和生物分类丰度方面,我们的方法比单端或双端方法具有更好的性能,这是由于在双端读取中充分利用了信息。
根据16S扩增子的大小和配对末端读段的长度,我们可能会有重叠或不重叠的配对末端reads。例如,对V4区域(252 nt,515F / 806R引物)进行测序可产生重叠的配对末端读数,而对V3-V5区域(694 nt,F357 / R926引物)进行测序可导致使用Illumina产生不重叠的PE reads MiSeq(250 bp×2)。由于QIIME和mothur当前不支持基于非重叠PE reads的从头OTU拣选,因此我们管道的主要优势在于能够处理非重叠配对末端读取。但是,我们的流程也可以通过使用PANDAseq 将成对的末端读取拼接在一起而应用于重叠的成对末端读取。要注意的是,一些现有的流水线还可以处理具有不同容量的双端和单端读取的混合。例如,最近提出的LotuS流程使用高质量的R1读取来构建OTU,然后对R1和R2进行后聚类合并以提高分类法的准确性。 但是,OTU级别的分辨率仍然由R1读取确定。
有一些针对16S数据开发的新管道。将这些先进的流程与Hybrid-denovo进行基准比较很有意思。我们选择DADA2和LotuS进行比较,因为它们已被证明比QIIME和mothur具有更好的整体性能,并且已被社区越来越多地使用。我们对具有完整读取对的金标准数据集重复了相同的分析。补充说明1中记录了用于DADA2和LotuS的特定命令行。DADA2产生18 389个序列变体(SV),而LotuS产生472个OTU。杂交denovo和LotuS之间的OTU / SV级Bray-Curtis距离上的Mantel相关性很高(ρ= 0.93),而杂交denovo和DADA2之间的Mantel相关性中等(ρ= 0.71)。有趣的是,所有方法之间在属水平Bray-Curtis距离上的Mantel相关性都很高(ρ> 0.97),这表明所有方法都可以产生相似的属水平分布(补充图4)。类似的ICC分析表明,所有方法均具有相对较高的ICC,但Hybrid-denovo总体上具有最佳性能(补充图5)。
从头OTU聚类的一个问题是潜在的OTU编号膨胀,这可能是由于诸如测序错误,嵌合体和环境污染物之类的来源引起的。 在Hybrid-denovo中,我们使用了各种质量过滤标准来减少虚假OTU的数量。 例如,我们应用Trimmomatic [4]修剪和删除了低碱基质量的读段,删除了含歧义碱基的读段,删除了单例OTU,使用Infernal软件包[9]删除了非结构对齐的OTU,并使用了基于参考的 UCHIME作为附加的嵌合体去除方法[6]。 但是,由于未知的排序错误,即使是这些滤波器也可能无法减少夸大的分集估计。 改进流程的多样性估计将是我们未来工作的重点。
(全文完)
2.再来看看这篇有同行不同意的文章
这篇文章是用于不同文章meta分析的,并不能用于非重叠reads序列的数据分析,反而是上面提到的两个流程中的LotuS流程可以尝试体验一下。摘要也放在这。
摘要
背景
对16S核糖体RNA(rRNA)基因的短而高变部分进行大规模高通量测序已改变了描述复杂生物群落内部和整个微生物群落中微生物多样性的方法论视野。但是,一些研究表明,所观察到的样品组成和分布是由方法而不是生物学差异引起的。尽管经常忽略这一事实,但这会损害荟萃分析。
结果
由于16S rRNA区域特异性的表现,通常观察到不同的生物学结果。 NG-Tax表现出了对区域选择和其他与16S rRNA基因扩增子测序研究相关的技术偏见的强大鲁棒性,从而减小了其影响并提供了真实样品组成的准确定性和定量表示。这将改善研究之间的可比性,并促进标准化工作。估计也是和上面一样的情况。QIIME作为常用管道。通过在Illumina的HiSeq2000平台上进行三个独立的测序运行,对两个可变的16S rRNA基因区域V4和V5-V6进行测序,对49个独立扩增的模拟样品的微生物组成进行了表征。这可以评估分类学分类中技术偏见的重要原因:1)逐次测序变异,2)PCR错误和3)区域/引物特异性扩增偏倚。尽管阅读时间短(〜140 nt)和所有技术偏见,但模拟群体中所包含系统型的分类学分配的平均特异性为97.78%。平均而言,至少有99.95%和88.43%的读数可以分配给家族或属,而“虚假属”的分配平均仅占每个样本读数的0.21%。对α和β多样性的分析证实了由生物学指导而不是上述方法论指导的结论,而QIIME未能实现这一结论。
结论
由于16S rRNA区域特异性的表现,通常观察到不同的生物学结果。 NG-Tax表现出了对区域选择和其他与16S rRNA基因扩增子测序研究相关的技术偏见的强大鲁棒性,从而减小了其影响并提供了真实样品组成的准确定性和定量表示。这将改善研究之间的可比性,并促进标准化工作
参考:
1.https://www.researchgate.net/post/do_you_know_the_possibilities_of_the_use_of_non-overlapping_Illumina_PE_reads_for_16S_rDNA_microbial_population_studies
3.NG-Tax, a highly accurate and validated pipeline for analysis of 16S rRNA amplicons from complex biomes