1.导入qiime2前的准备
我简单处理了下otu序列和表,使它们能导入qiime2,应该是一行shell代码解决的,shell水平不行,python来顶了。
import os
fout = open('otus.txt', 'w')
fout_otab = open('otab.txt', 'w')
with open('dada2_counts.txt') as f:
i = 1
for line in f:
if i == 1:
fout_otab.write('#OTU\t' + line.split('\t')[0])
i += 1
continue
seq_name = '>OTU' + str(i)
fout.write(seq_name +'\n')
number = line.split('\t')[1]
seq = line.split('\t')[0]
fout.write(seq + '\n')
fout_otab.write(seq_name.split('>')[1] + '\t' + number)
i += 1
fout.close()
fout_otab.close()
2.qiime流程走一回
基本上是参考官方文档和宏基因组微信公众号的,
#转成biom格式
biom convert -i otab.txt -o otab.biom --table-type="OTU table" --to-json
#导入ASV表
qiime tools import \
--input-path otab.biom \
--type 'FeatureTable[Frequency]' \
--input-format BIOMV100Format \
--output-path table.qza
# 导入参考序列
qiime tools import \
--type 'FeatureData[Sequence]' \
--input-path otus.fasta \
--output-path otus.qza
echo 统计并可视化
qiime feature-table summarize \
--i-table table.qza \
--o-visualization table.qzv
echo 代表序列统计并可视化
qiime feature-table tabulate-seqs \
--i-data otus.qza \
--o-visualization otus.qzv
来看看序列,因为前面已经处理好了,这里应该是没有变化的,只是个统计而已。
 ]
]
echo 分类 Taxonomic analysis
qiime feature-classifier classify-sklearn \
--i-classifier gg-13-8-99-515-806-nb-classifier.qza\
--i-reads otus.qza \
--o-classification taxonomy.qza
echo 可视化
qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
# 准备sample--metadata.tsv然后画个柱状图
echo 画图
qiime taxa barplot \
--i-table table-with-phyla.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization taxa-bar-plots-p.qzv
这里就获得了一个柱状图,虽然只的一个样本,
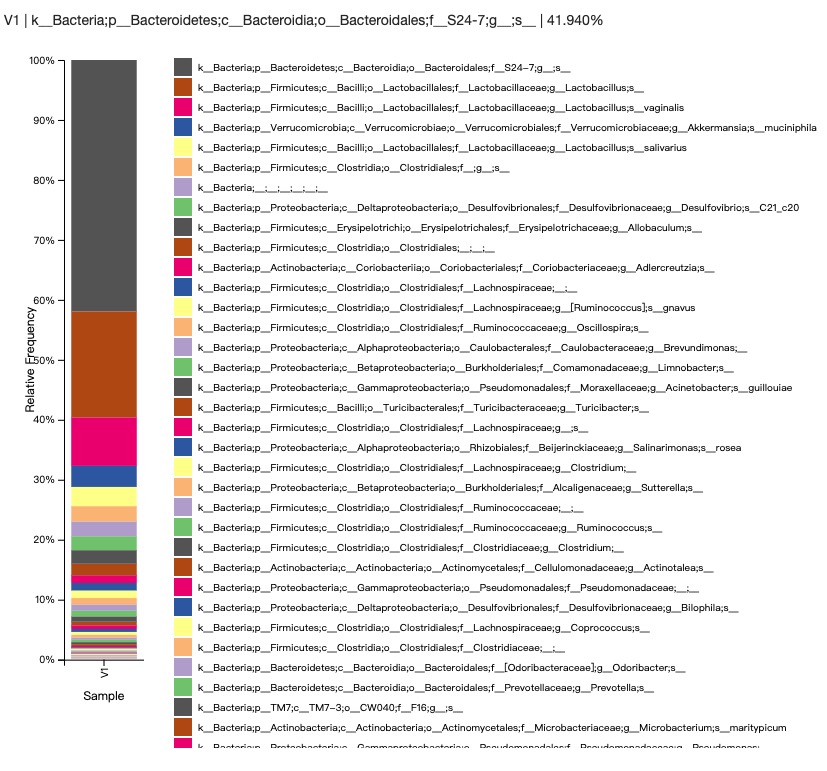
那么现在这条路走通了,后面做批量样本就可以了,只是官方不推荐这么做的,毕竟,不确定这样做会有多少物种错误分类或者假阳性。还需要后面的验证,比如跑个模拟菌落,能不能把数据库优化成和测序数据拼接后一样的,这样是不是准确度会更高一点。定制流程是一个比较有难度的活。
qiime2的简单探索暂时就到这里,后面继续接着用两个R包进行物种注释看看结果,方便的话比较一下两者的差别。然后学习一下phyloseq进行分析,这个包好像也比较有名,基本上还是官方文档的小参数修改而已。
前面已经进行的探索有: