继续之前未完成的笔记,前面实现了使用qiime2-dada2插件初步探索,结果以无敌的报错失败告终,这里进入R包,更灵活地处理数据,下面是我的详细步骤。
1.上次未完成的准备工作
#设置R包的工作目录,不是R语言的,这只是个变量而已
path <- "/Users/zd200572/Biodata/16S/process-ubiome-16S/primer-cutted"
# 获得目录中的数据列表
fnFs <- sort(list.files(path, pattern="_1.fastq", full.names = TRUE))
fnRs <- sort(list.files(path, pattern="_2.fastq", full.names = TRUE))
# 获取样本名称
sample.names <- sapply(strsplit(basename(fnFs), "_"), `[`, 1)
#看下样本,为了简单,我就放了一个样本
fnFs
[1] "/Users/zd200572/Biodata/16S/process-ubiome-16S/primer-cutted/sIL10-2-S-4_1.fastq"
2.数据质量质控
质量控制是必由之路,看下这个质量图,与qiime2的交互动态图不同,这个是静态的,
#看看质量情况,可以看出最后的质量值差到不忍直视了,难怪拼接率只有3%
plotQualityProfile(fnFs[1:2]) #正向质量
plotQualityProfile(fnRs[1:2]) #反向质量


3.过滤和修剪
这里还有个严重不合格样本的数据过滤,为了流程命名不变,我也走一遍。
# Place filtered files in filtered/ subdirectory
filtFs <- file.path(path, "filtered", paste0(sample.names, "_F_filt.fastq.gz"))
filtRs <- file.path(path, "filtered", paste0(sample.names, "_R_filt.fastq.gz"))
names(filtFs) <- sample.names
names(filtRs) <- sample.names
然后,我又通过fastqc看了下,序列基本上都是在120bp以后质量显著下降,于是正反向从120bp开始截取。
out <- filterAndTrim(fnFs, filtFs, fnRs, filtRs, truncLen=c(120,120),
maxN=0, maxEE=c(2,2), truncQ=2, rm.phix=TRUE,
compress=TRUE, multithread=TRUE) # On Windows set multithread=FALSE
head(out)
reads.in reads.out
sIL10-2-S-4_1.fastq 198387 194949
errF <- learnErrors(filtFs, multithread=TRUE)
errR <- learnErrors(filtRs, multithread=TRUE)
plotErrors(errF, nominalQ=TRUE)
这里说明一下,如果用之前我说的github上ubiome里面的数据的话,可能还要按照具体情况具体分析,应该是已经质控好的,直接加N即可,不过估计切到120再做也可以,就是损失几个碱基而已。
4.训练错误率模型?
因为我是一个样本,速度还是挺快的,几乎是秒完成。i3-3120M处理器,比较老啦,凑和还能用两年。然后做下来结果拟合度对比官方的感觉还挺好。

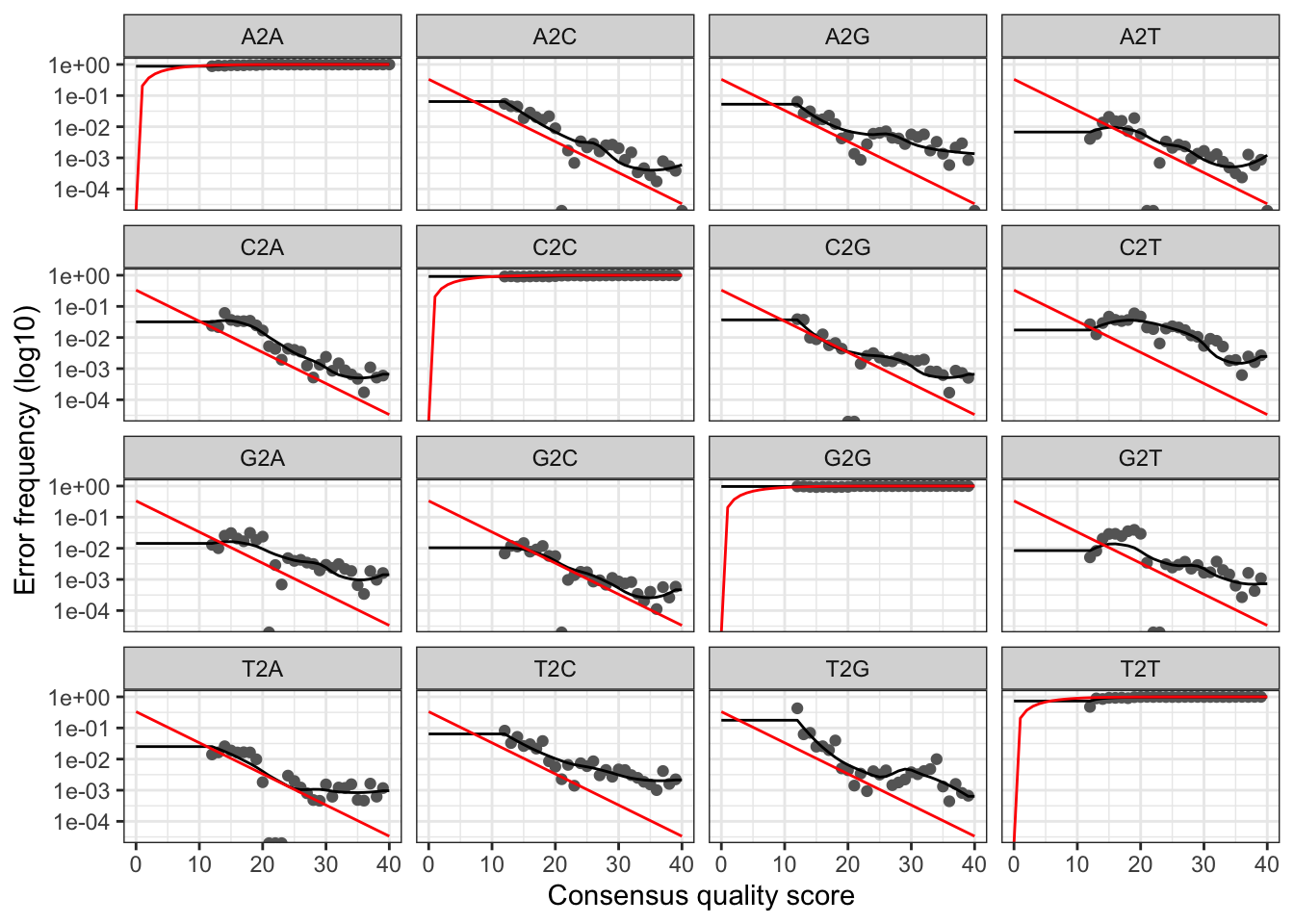
官方的,区别不大吧,所以应该是可以用的,直接继续了。
5.样本自参考
这应该是把上面建好的模型应用于我们的序列,这一步还是比较快的,也可能我只有一个样本的原因。作者提示如果windows系统要把multithread设置为FALSE。
dadaFs <- dada(filtFs, err=errF, multithread=TRUE)
#Sample 1 - 194949 reads in 11507 unique sequences.
dadaRs <- dada(filtRs, err=errR, multithread=TRUE)
#Sample 1 - 194949 reads in 11385 unique sequences.
dadaFs[[1]]
TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGCGAGTTAAGTCAGCGGTAAAAGCCCGGGGCTCAACCCCGGCCCGCCGTTGAAACTGGCTGGCTTG 35756
6.合并双向reads
这里就是最最关键合并步骤了,justConcatenate=TRUE把序列直接以10个N拼接,亮眼的10个N。
mergers <- mergePairs(dadaFs, filtFs, dadaRs, filtRs, verbose=TRUE, justConcatenate=TRUE)
head(mergers[[1]]) #这里可以很明显地看到10个N
$TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGCGAGTTAAGTCAGCGGTAAAAGCCCGGGGCTCAACCCCGGCCCGCCGTTGAAACTGGCTGGCTTG
[1] "TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGCGAGTTAAGTCAGCGGTAAAAGCCCGGGGCTCAACCCCGGCCCGCCGTTGAAACTGGCTGGCTTGNNNNNNNNNNGCAGGCGGAATGCGCGGTGTAGCGGTGAAATGCATAGATATCGCGCAGAACCCCGATTGCGAAGGCAGCCTGCCGGCCCCACACTGACGCTGAGGCACGAAAGCGTGGGTATCGAACAGG"
7.构建序列表
这就是当年的OTU表,现在流行的ASV featuretable了,这里与之前OTU的区别应该是相似度,现在都是以100%相似度聚类了。
seqtab <- makeSequenceTable(mergers)
dim(seqtab)
[1] 1 500
8.去嵌合
去嵌合步骤中,作者提示如果大部分样本被除去,或者序列长度不对,要运行一下注释的那句。
table(nchar(getSequences(seqtab)))
#seqtab2 <- seqtab[,nchar(colnames(seqtab)) %in% 250:256]
seqtab.nochim <- removeBimeraDenovo(seqtab, method="consensus", multithread=TRUE, verbose=TRUE)
head(seqtab.nochim)
# TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGCGAGTTAAGTCAGCGGTAAAAGCCCGGGGCTCAACCCCGGCCCGCCGTTGAAACTGGCTGGCTTGNNNNNNNNNNGCAGGCGGAATGCGCGGTGTAGCGGTGAAATGCATAGATATCGCGCAGAACCCCGATTGCGAAGGCAGCCTGCCGGCCCCACACTGACGCTGAGGCACGAAAGCGTGGGTATCGAACAGG
#[1,] 35670
sum(seqtab.nochim)/sum(seqtab)
[1] 0.9975397
9.最后的检查
这里相当于汇总一下序列从原始到各个步骤,去除了多少条。
getN <- function(x) sum(getUniques(x))
track <- cbind(out, getN(dadaFs), getN(dadaRs), getN(mergers), rowSums(seqtab.nochim))
colnames(track) <- c("input", "filtered", "denoisedF", "denoisedR", "merged", "nonchim")
rownames(track) <- sample.names
head(track)
10.物种注释
#下载数据库,第一次运行
#download.file("https://zenodo.org/record/158955/files/gg_13_8_train_set_97.fa.gz?download=1",destfile = 'gg_13_8_train_set_97.fa.gz')
taxa <- assignTaxonomy(seqtab.nochim, "gg_13_8_train_set_97.fa.gz", multithread=TRUE)
#这个步骤不好用,可能是N太多导致不能更好分析到种,应该说对于这种盲目拼接的序列,到属是最好的选择,因为可能存在序列+10N之后拼接起来indel有多个。还可能是greengenes本身这个训练集不能到种,需要换另外的RDP或者Silva,以后再处理这个问题,而且后面我选择使用qiime2进行物种注释,发现这样结果更好些。
#taxas <- addSpecies(taxa, "/Users/zd200572/Biodata/16S/process-ubiome-16S/primer-cutted/rdp_species_assignment_16.fa.gz")
#Error in .Call2("ACtree2_build", tb, pp_exclude, base_codes, nodebuf_ptr, :
# non base DNA letter found in Trusted Band for pattern 1
taxa.print <- taxa # Removing sequence rownames for display only
rownames(taxa.print) <- NULL
head(taxa.print)[1,]
Kingdom Phylum Class Order
"k__Bacteria" "p__Bacteroidetes" "c__Bacteroidia" "o__Bacteroidales"
Family Genus Species
"f__S24-7" "g__" "s__"
#虽然这个只注释到科,还是可以接受的,毕竟v4只有1/8 16S全长序列。
12.导出序列
因为我R语言水平不咋的,就直接使用刘大哥的代码了:
#保存导出文件
setwd(path)
seqtable.taxa.plus <- cbind('#seq'=rownames(taxa), t(seqtab.nochim), taxa)
# ASV表格导出
write.table(t(seqtab.nochim), "dada2_counts.txt", sep="\t", quote=F, row.names = T)
# 带注释文件的ASV表格导出
write.table(seqtable.taxa.plus , "dada2_counts.taxon.species.txt", sep="\t", quote=F, row.names = F)
# track文件保存
write.table(track , "dada2_track.txt", sep="\t", quote=F, row.names = F)
#————————————————
#版权声明:本文为CSDN博主「刘永鑫Adam」的原创文章,遵循 CC 4.0 BY-SA 版权协议,转载请附上原文出处链接及本声明。
#原文链接:https://blog.csdn.net/woodcorpse/article/details/86773312