一直迷惑于如何把qiime2和picrust结合起来用来分析16S的数据,直到这两天,看到了微生太公众号的视频教程,才有了眉目,原来如此。详细视频教程可以查找相关公众号获得。前面看到picrust2已经处于beta状态了,其可以嵌入于qiime2中,使用更方便,可是我的试用结果却差强人意,或许是我的使用过程有问题,16G内存的要求一般的电脑也难以实现。之前使用picrust1网页版(Galaxy平台,不是三星的那个,是个生物信息云平台软件系统)分析的效果还可以,于是决定用picrust1再试试。发现pcirust在今年6月份更新了1.1.4版。
再来吐槽几句这个功能预测,虽然大部分预测准确率挺高,还是不能说明真正的问题,毕竟真实的宏基因组才是最准的,随着测序成本的降低,功能预测有一天也许会成为历史。地球上人与人之间的DNA的差别在0.03%(算上SNP,CNV和indel等等),而细菌菌属间的差别远比这差得多(由于基因间的水平转移等),所以只是参考。
刚刚搜索了一下,PICRUSt2已经于预印版发布了,6月份发布的,文章地址在这,阅读原文,也可以看到文章。晚会再学习一下这篇文章。
一、所需文件下载
这里我是直接使用qiime2官方教程里的文件,(一不小心又发现qiime2也更新了,软件更新可真快呀!)https://docs.qiime2.org/2019.7/data/tutorials/otu-clustering/table.qza , https://docs.qiime2.org/2019.7/data/tutorials/otu-clustering/rep-seqs.qza , ftp://greengenes.microbio.me/greengenes_release/gg_13_5/gg_13_5_otus.tar.gz 参考视频教程的脚本里的命令来完成的。脚本里是根据他们商业化报告来处理的,这里我们是直接从qiime2分析我是来的数据,所以基本不需要文件导入qiime2这一操作了,otu参考数据还是需要导入一下的。
二、处理成picrust1所需
#解压greengenes-13-5参考数据库
tar zxvf _otus.tar.gz
#找到这个文件gg_13_5_otus/rep_set97_otus.fasta,可以把它放在工作目录,这就是我们要使用的参考序列
#因为一般只是测16s-V4区,所以常规分析绝大部分只能到属,所以选择97%相似度
#安装好qiime2,然后导入成qiime2格式,方便后面使用
qiime tools import --input-path ../97_otus.fasta --output-path ref-sequences.qza --type 'FeatureData[Sequence]'
qiime vsearch cluster-features-closed-reference --i-sequences rep-seqs.qza --output-dir closed-ref-otu/ --i-table table.qza --i-reference-sequences ref-sequences.qza --p-perc-identity 0.97
qiime tools export --input-path closed-ref-otu/clustered_table.qza --output-path closed-ref-otu/
biom convert --to-tsv -i closed-ref-otu/feature-table.biom -o closed_reference_otu_table.tsv
三、使用网页版软件或者手动安装
使用网页版的就很简单了,只是手动点点就好了,上传biom或者tsv格式。下面是我的软件安装和使用步骤,软件安装参考官方教程,使用参考宏基因组微信公众号。
1.软件安装
#使用已经安装好的miniconda安装,简便快捷
conda create -n picrust1 -c bioconda -c conda-forge picrust
#下载所需数据库
wget http://kronos.pharmacology.dal.ca/public_files/picrust/picrust_precalculated_v1.1.4/13_5/ko_13_5_precalculated.tab.gz
wget http://kronos.pharmacology.dal.ca/public_files/picrust/picrust_precalculated_v1.1.4/13_5/ko_13_5_precalculated.tab.gz
#建立data文件夹
mkdir -p /Volumes/MacOS/Miniconda3/envs/picrust1/lib/python2.7/site-packages/picrust/data
#复制数据库文件到文件夹,不需要解压
cp ko_13_5_precalculated.tab.gz ko_13_5_precalculated.tab.gz /Volumes/MacOS/Miniconda3/envs/picrust1/lib/python2.7/site-packages/picrust/data
#激活环境
conda activte picrust1
2.软件使用
#按拷贝数标准化OTU表
normalize_by_copy_number.py -i closed-ref-otu/feature-table.biom -o normalized_otus.biom
#预测宏基因组
predict_metagenomes.py -i normalized_otus.biom -o metagenome_predictions.biom
# 转换格式为txt
biom convert -i metagenome_predictions.biom -o metagenome_predictions.txt --table-type="OTU table" --to-tsv
#调整为标准表格
#sed -i '/# Const/d;s/#OTU //g' metagenome_predictions.txt 我是黑苹果,可能mac sed命令有所差别,没有#完成这个命令。
#按功能级别分类汇总,-c指输出类型,有KEGG_Pathways, COG_Category,RFAM三种,-l是级别,分3级
categorize_by_function.py -i metagenome_predictions.biom -c KEGG_Pathways -l 3 -o metagenome_predictions.L3.biom
# 转换格式为txt,然后就可以看结果了,使用excel打开即可清楚查看。
biom convert -i metagenome_predictions.L3.biom -o metagenome_predictions.L3.txt --table-type="OTU table" --to-tsv
#同样没有完成 sed -i '/# Const/d;s/#OTU //g' metagenome_predictions.L3.txt
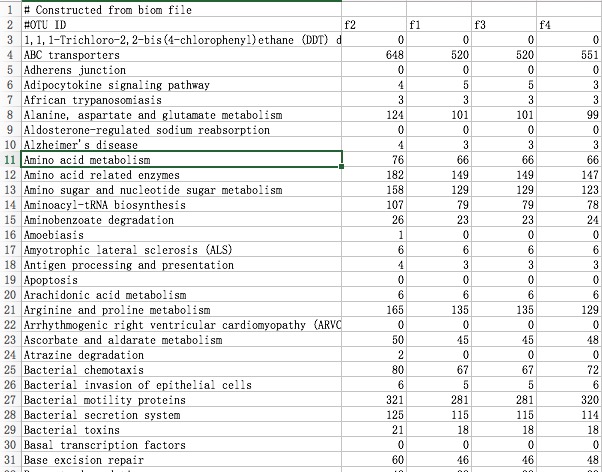
3.看看结果
这就是美美的结果了,和宏基因组一样可以做许多额外分析了。