身处这样一个互联网时代,应当感恩技术带来的便利,从在一个地方不远游就只能是井底之蛙,到今天互联网让我们不出门知天下事,当然,假消息也有。虽然现在许多事和技能仍然需要项目实践,但是不得不说,知识已经不再是一种稀缺的资源,需要时间训练的技能才是。我们应该充分利用好这个时代提供给我们的便利,努力学习和思考。
虽然川普四处设限,但是地球村依然变得越来越“小”,就拿我们生命科学领域来说,ncbi数据库,让我们每个人都有机会接触到测序原始数据,可以进行分析再现和学习。手上虽然没有“便宜”的纳米孔测序仪,但是借助科学研究者的数据,依然可以对其一探究竟。这里,我在牛津纳米孔公司官网看到了几篇最新发表的采用其技术浓度测序16S的文献,下载了原始数据,学习一下测16S的可行性和数据分析方法。
令我大跌眼镜的原始数据
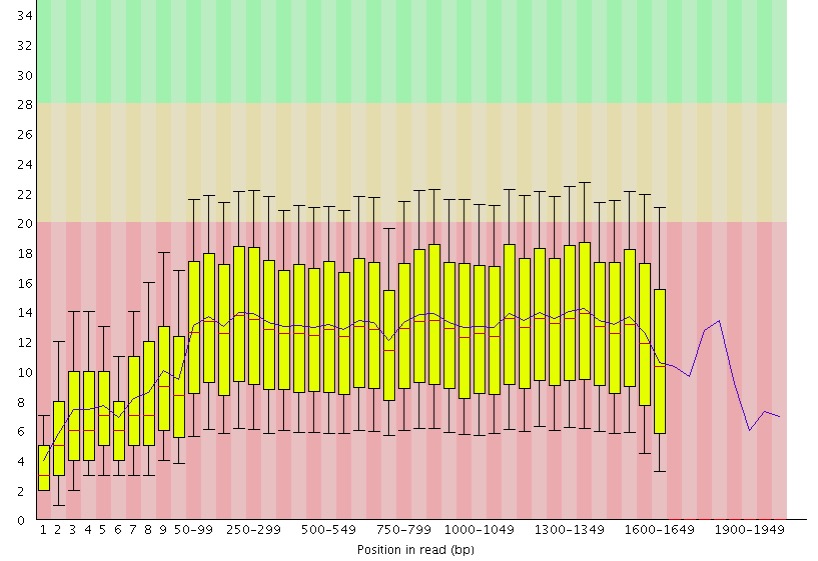
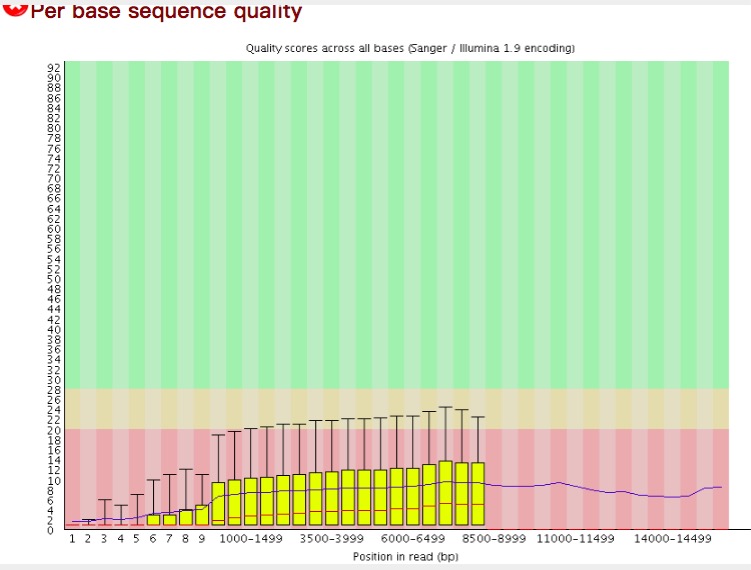
随便拿了几个数据,fastqc来看一下,好家伙,质量确实有点低,当然,这应该是R9.4,9.5或者更早版本的试剂,相信以后会更好。看来直接测了分析高可变区的16S是不怎么可行的,当然,如果有特殊方法来解决是可以的,比如Pacbio的循环测序和把一个拷贝多份连在一条上,也实现测多次的效果,当然,依然无法消除那种系统错误,比如技术本身缺陷,插入或缺失(后面的NanoApli-seq就是后面一种方法)。还不得不吐槽一下这家公司,只对有测序仪的用户开放社区论坛,这样就让技术只局限在了一个小圈子,封闭并不利于该公司的发展。
几篇文章的略读
- 1.Cuscó A, Catozzi C, Viñes J et al. Microbiota profiling with long amplicons using Nanopore sequencing: full-length 16S rRNA gene and whole rrn operon
这篇文章采用了比较测16S和rrn序列(16S rRNA–ITS–23S rRNA; 4,500 bp),结果使用EPI2ME的话16S序列中只有68%的序列能够匹配到正确的分类。我学得这个方法基本上没有可用性呢。

- 2.E. Curren, T. Yoshida, V.S. Kuwahara et al. Rapid profiling of tropical marine cyanobacterial communities
- 3.Rapid bacterial identification by direct PCR amplification of 16S rRNA genes using the MinION nanopore sequencer
- 4.NanoAmpli-Seq: a work ow for amplicon sequencing for mixed microbial communities on the nanopore sequencing platform
这篇文章采用9.4版本的试剂,1D的建库方式,得到的平均Q值为11.7,算了下准确度为91.17%,大概也就这么高了。这篇文章是采用qiime流程进行后续处理的。这篇文章是测热带海洋蓝藻的,对于细菌菌落可能不大能说明问题。
这篇文章的流程如下图所示:
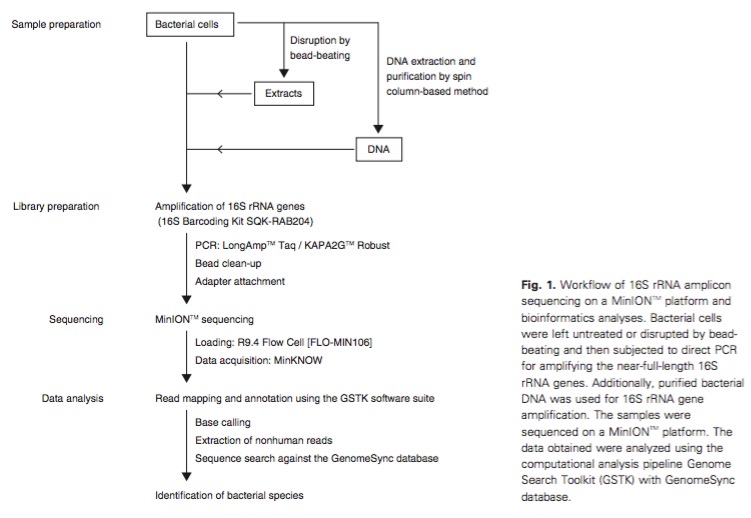
是使用 GSTK software suite进行数据分析的(比对和注释序列)。
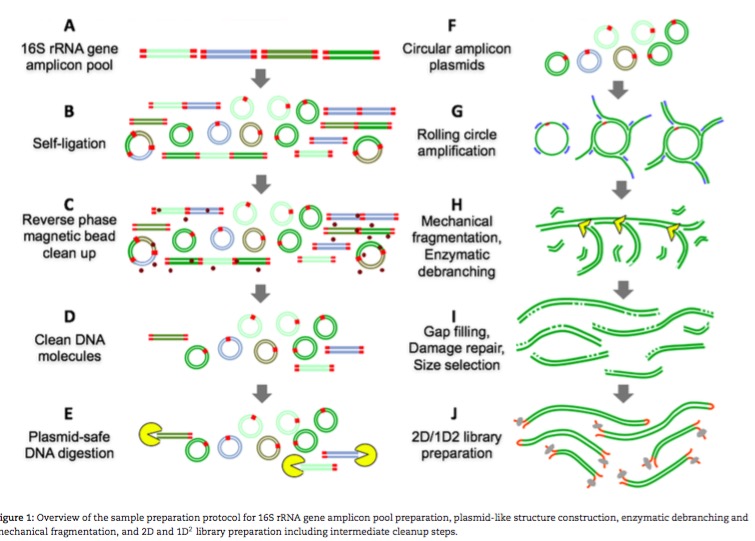
这篇是我前面提到的采用串联线性片段进行测序的文章,看它的文库制备有些复杂,原理图放在这:
最后一篇文章分析过程学习
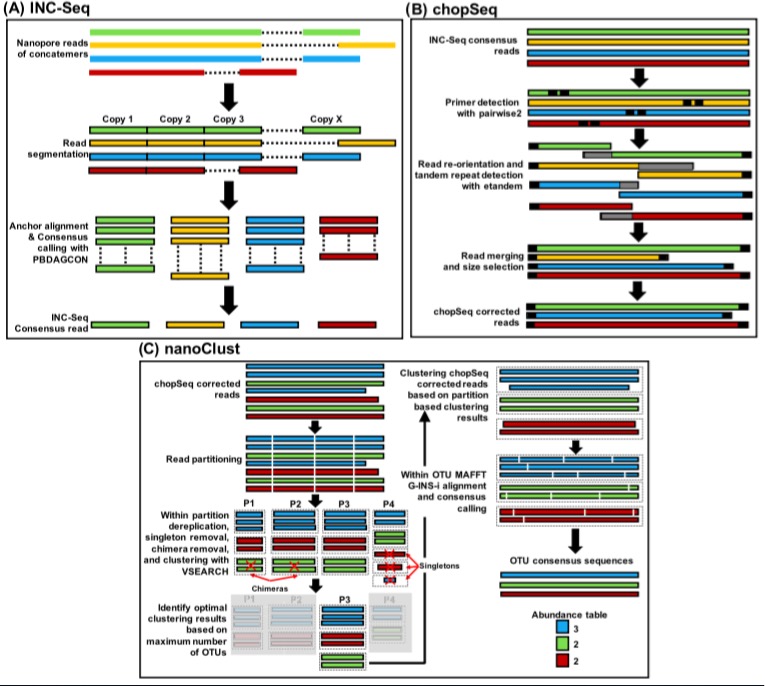
上面这张图是关于数据分析的过程图解,主要包括INC-Seq,ChaoSeq, nanoClust三个过程,后两个分别对应了两个脚本文件chopSEQ.py和nanoCLUST.py。第一个应该是整个过程的预览。作者公开了两个数据,能下载的只有一个,ERR2241540.sra,大小是10M,fasq-dump解压完只有4.6M,我感到很意外,压缩压大了?查了下,还真有这种情况出现。
看到讨论里的几句话,瞬间觉得纳米孔不适合做这种16S群落分析,特别是物种组成复杂时。
1.由于序列质量不够,没办法使用vsearch等软件进行聚类,只能通过分区序列聚类来基本满足物种分类要求;
2.150X, 也就是50个长reads(3X),可以实现共识序列精度达到99%+。但是精度仍然低于illumina或者Pacbio的测序准确度(Pacbio不是系统错误,是随机错误)。而且,即使增加测序深度,精度也不会提高,这说明至少在现阶段,这的确是个系统错误;
3.产量低,能basecalling的仅仅是原始数据的一小部分,如7%–9%的1D方数据。如果使用1D的建库方式或许能解决这个问题,但是精度只有94%,就不适合进行上述的聚类了;
4.一个聚类会产生多个共识序列,可能会导致物种分类错误。
如果有可能的话,后面学习一下它的分析过程命令行,现在卡在了软件安装上,晚会续上。