最近在github上找到一个ubiome的原始数据,包含多个身体部位的,尝试分析一下,看看能获得什么结果。经历了那么多的安装坑,发现还是docker的安装方式最方便有效、节省时间,特别是换上国内的加速源之后。虚拟机太占资源,而且总感觉虚拟机不够真实,conda安装完竟然也总是报错,特别是2个月更新一次,如此频繁的qiime2。
1.docker安装qiime2
#安装docker
sudo apt-get install \
apt-transport-https \
ca-certificates \
curl \
gnupg2 \
software-properties-common
curl -fsSL https://download.docker.com/linux/debian/gpg | sudo apt-key add -
sudo add-apt-repository \
"deb [arch=amd64] https://download.docker.com/linux/ubuntu \
$(lsb_release -cs) \
stable"
sudo apt-get update
sudo apt-get install docker-ce
#更新国内DaoCloud 加速源,看教程也有阿里云的
https://www.daocloud.io/mirror#accelerator-doc
##将用户添加到docker组 ,方便使用
#非root用户,不需要使用sudo使用docker,需要将用户添加到docker组sudo groupadd docker
sudo gpasswd -a 用户名 docker
sudo service docker restart
#摘取qiime镜像
sudo docker pull qiime2/core
sudo docker run -t -i --rm -v $(pwd):/data qiime2/core
#这里我们在docker里的工作目录就变成了/data,然后数据的绝对目录就是/data/microbiome
2.原始数据的稍微处理
由于ubiome的测序数据是双向150bp,去除引物得到的单向有效长度是120bp多,而一般V4全长是250bp左右,双向数据是难以拼接的,所以这里就简单把两端数据混在一起分析了,不知道科不科学。ubiome是采用完全比对到SILVA数据库,然后选择完全比对的序列作分析的,这个分析方法还是申请了专利的,有点复杂,这里就按简单的来了。当然,一般分析16S数据是要切去扩增引物的,这里为了简单也省去了。
#首先,下载这些数据: git clone https://github.com/gedankenstuecke/microbiome.git cd microbiome #这里面有两次的肠道菌检测数据,把最近更新的一次数据(SSR_23172 - gut 2 (taken on September 5th, 2015))的正反向文件合并 zcat ssr_23172__R1* >> ssr_23172.fastq zcat ssr_23172__R2* >> ssr_23172.fastq gzip ssr_23172.fastq mkdir raw_data mv ssr && mv ssr_23172.fastq.gz raw_data/
3.导入数据
#把数据重新命名成qiime2能识别的命名方式,比如XX_S67_L001_R2_001.fastq,这样直接导入更方便些: qiime tools import \ --type 'SampleData[SequencesWithQuality]' \ --input-path raw_data \ #你的重命名数据所在的文件夹 --input-format CasavaOneEightSingleLanePerSampleDirFmt \ --output-path demux-single-end.qza
4.序列质控及Feature表构建
## quality control #visualization qiime demux summarize \ --i-data microbiome.qza\ --o-visualization microbiome.qzv ##filter 聚类 qiime dada2 denoise-single \ --i-demultiplexed-seqs microbiome.qza \ --p-trunc-left 0 \ --p-trunc-len 126 \ --o-representative-sequences rep-seqs-dada2.qza #输出feature表代表序列 --o-table table-dada2.qza \ #输出表 --p-n-threads 4 \#4线程,服务器是36线程的 --o-denoising-stats stats-dada2.qza #qzv 可视化 qiime feature-table summarize \ --i-table table-dada2.qza \ --o-visualization table-dada2.qzv #代表序列统计 qiime feature-table tabulate-seqs \ --i-data rep-seqs-dada2.qza \ --o-visualization rep-seqs.qzv qiime tools view rep-seqs.qzv
5.建树,用于多样性分析
# 多序列比对 qiime alignment mafft \ --i-sequences rep-seqs.qza \ --o-alignment aligned-rep-seqs.qza # 移除高变区 qiime alignment mask \ --i-alignment aligned-rep-seqs.qza \ --o-masked-alignment masked-aligned-rep-seqs.qza # 建树 qiime phylogeny fasttree \ --i-alignment masked-aligned-rep-seqs.qza \ --o-tree unrooted-tree.qza # 无根树转换为有根树 qiime phylogeny midpoint-root \ --i-tree unrooted-tree.qza \ --o-rooted-tree rooted-tree.qza
6.alpha多样性
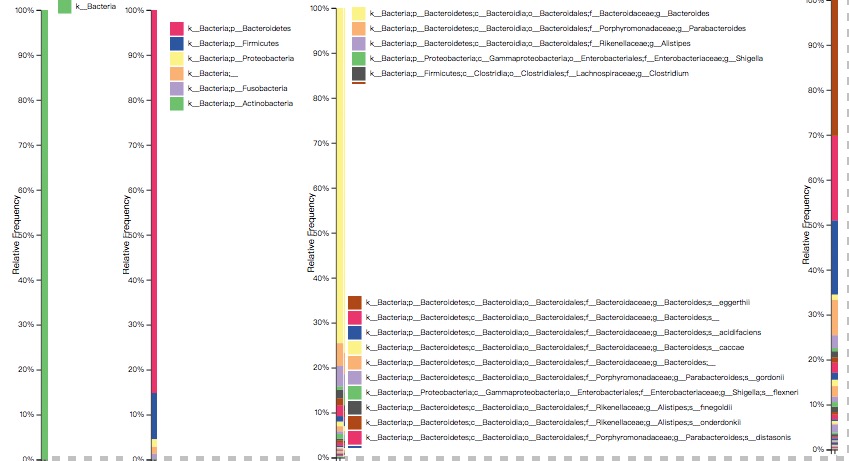
qiime diversity core-metrics \ --i-phylogeny rooted-tree.qza \ --i-table table.qza \ --p-sampling-depth 1080 \ --output-dir core-metrics-results #物种注释,greengenes13.8 V4区数据注释 qiime feature-classifier classify-sklearn \ --i-classifier gg-13-8-99-515-806-nb-classifier.qza \ --i-reads rep-seqs-dada2.qza \ --o-classification taxonomy.qza #可视化看看 qiime metadata tabulate \ --m-input-file taxonomy.qza \ --o-visualization taxonomy.qzv #画个柱状图,一般人最想要的就是这个图吧 qiime taxa barplot \ --i-table table-dada2.qza \ --i-taxonomy taxonomy.qza \ --m-metadata-file sample-metadata.tsv \ --o-visualization taxa-bar-plots.qzv
最后就得到这张漂亮的图啦,我截取了界、门、属和种四个分类级别的柱状图。