软件简介
 丹麦研究团队的Hof和Speed推出了一款名为LDAK-KVIK的新工具,旨在解决当前全基因组关联研究(GWAS)领域的一个核心痛点。目前,混合线性模型(MMAA)已成为GWAS分析的首选方法,因为它能有效控制群体结构和亲缘关系造成的假阳性,并提高统计功效。然而,研究者们常常不得不在计算速度和分析效能之间做出艰难的权衡。LDAK-KVIK的出现,似乎为我们提供了一个两全其美的解决方案,它在保证顶尖统计功效的同时,还具备极高的计算效率。
丹麦研究团队的Hof和Speed推出了一款名为LDAK-KVIK的新工具,旨在解决当前全基因组关联研究(GWAS)领域的一个核心痛点。目前,混合线性模型(MMAA)已成为GWAS分析的首选方法,因为它能有效控制群体结构和亲缘关系造成的假阳性,并提高统计功效。然而,研究者们常常不得不在计算速度和分析效能之间做出艰难的权衡。LDAK-KVIK的出现,似乎为我们提供了一个两全其美的解决方案,它在保证顶尖统计功效的同时,还具备极高的计算效率。 该工具的强大功效,主要源于其对遗传结构更精细、更符合生物学实际的建模方式。传统的MMAA工具,如BOLT-LMM和REGENIE,通常假设所有单核苷酸多态性(SNP)的遗传力贡献是均等的,即遗传力与其次要等位基因频率(MAF)的关系固定。LDAK-KVIK打破了这一常规,它能够根据实际数据,灵活地估计并模拟每个SNP的遗传力如何随MAF变化。此外,它还采用了一种包含高斯分布和拉普拉斯分布的混合先验(elastic net prior),这比传统方法单一的高斯分布假设更能捕捉复杂的SNP效应量分布。这些模型的优化,使得LDAK-KVIK在分析的第一步就能构建出更精准的留一染色体排除(LOCO)多基因评分(PGS),而这正是提升第二步关联检验效能的关键所在。
该工具的强大功效,主要源于其对遗传结构更精细、更符合生物学实际的建模方式。传统的MMAA工具,如BOLT-LMM和REGENIE,通常假设所有单核苷酸多态性(SNP)的遗传力贡献是均等的,即遗传力与其次要等位基因频率(MAF)的关系固定。LDAK-KVIK打破了这一常规,它能够根据实际数据,灵活地估计并模拟每个SNP的遗传力如何随MAF变化。此外,它还采用了一种包含高斯分布和拉普拉斯分布的混合先验(elastic net prior),这比传统方法单一的高斯分布假设更能捕捉复杂的SNP效应量分布。这些模型的优化,使得LDAK-KVIK在分析的第一步就能构建出更精准的留一染色体排除(LOCO)多基因评分(PGS),而这正是提升第二步关联检验效能的关键所在。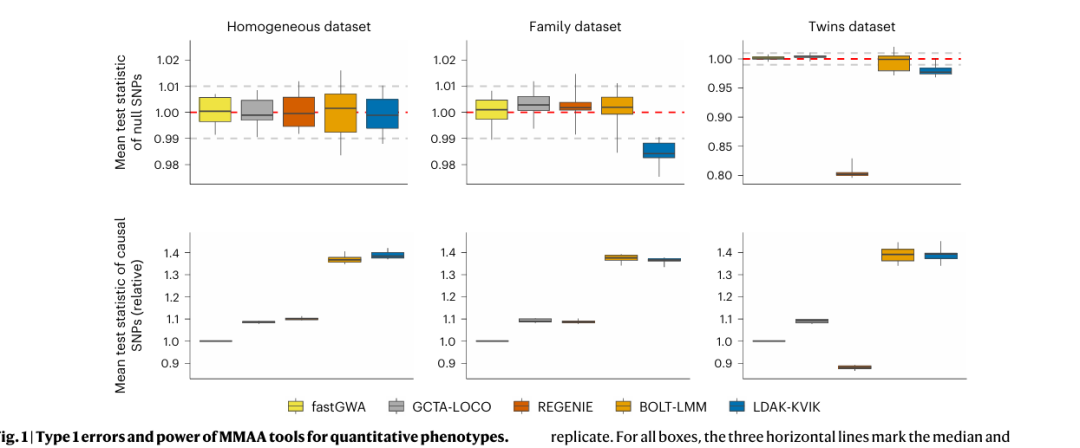 在计算效率方面,LDAK-KVIK也实现了重大突破。其核心创新在于开发了一种基于“区块”的变分贝叶斯求解器(chunk-based variational Bayes solver)。这种算法不需要一次性将所有基因型数据加载到内存中,而是分块处理,极大地降低了内存需求。更重要的是,相比传统的全基因组扫描更新方式,这种分块迭代收敛的策略所需的更新次数减少了5到20倍,从而显著缩短了运行时间。实际测试表明,分析一个包含约35万个体的大型数据集,LDAK-KVIK仅需不到10个CPU小时和5Gb内存即可完成,这一性能表现远超BOLT-LMM等工具,与以速度见长的REGENIE相比也毫不逊色。
在计算效率方面,LDAK-KVIK也实现了重大突破。其核心创新在于开发了一种基于“区块”的变分贝叶斯求解器(chunk-based variational Bayes solver)。这种算法不需要一次性将所有基因型数据加载到内存中,而是分块处理,极大地降低了内存需求。更重要的是,相比传统的全基因组扫描更新方式,这种分块迭代收敛的策略所需的更新次数减少了5到20倍,从而显著缩短了运行时间。实际测试表明,分析一个包含约35万个体的大型数据集,LDAK-KVIK仅需不到10个CPU小时和5Gb内存即可完成,这一性能表现远超BOLT-LMM等工具,与以速度见长的REGENIE相比也毫不逊色。
为了验证工具的可靠性与先进性,研究人员使用了模拟数据和来自英国生物样本库(UK Biobank)的大量真实表型数据,与当前主流的MMAA工具进行了全面的基准比较。在对40个UK Biobank定量表型的分析中,LDAK-KVIK的表现尤为突出,相较于传统的线性回归,它多发现了16%的独立全基因组显著位点,这一增幅超过了BOLT-LMM(15%)和REGENIE(11%)。在基因水平的关联分析中,其优势同样显著,发现的显著基因比现有领先工具多出18%。这些结果充分证明,LDAK-KVIK通过其创新的模型设计和算法优化,实实在在地提升了我们从大规模数据中发掘遗传信号的能力。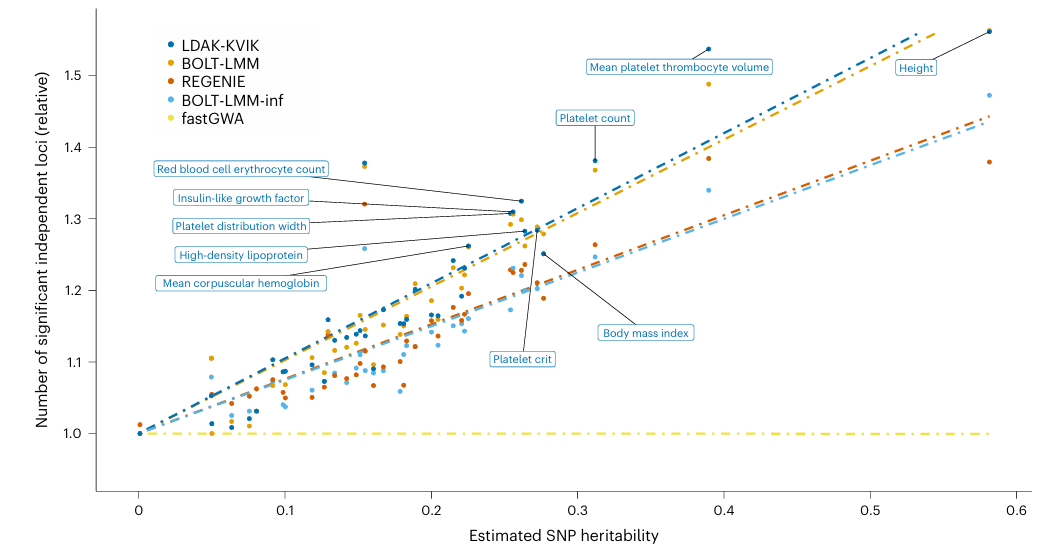 当然,研究也客观地指出了该工具的一些局限性。例如,在分析二元表型(如疾病状态)时,所有MMAA工具(包括LDAK-KVIK)的效能优势相比传统逻辑回归都不太明显,这主要是因为这类表型的遗传度较低,难以构建准确的PGS。尽管如此,LDAK-KVIK在各种复杂数据场景下,包括多种族混合数据和高度亲缘关系数据,都表现出了良好的I型错误控制能力,确保了结果的可靠性。总而言之,LDAK-KVIK的问世为GWAS领域提供了一个强大、高效且可靠的新选择,使研究者无需再为计算资源和分析深度而烦恼,有望推动复杂性状遗传机制的进一步解析。
当然,研究也客观地指出了该工具的一些局限性。例如,在分析二元表型(如疾病状态)时,所有MMAA工具(包括LDAK-KVIK)的效能优势相比传统逻辑回归都不太明显,这主要是因为这类表型的遗传度较低,难以构建准确的PGS。尽管如此,LDAK-KVIK在各种复杂数据场景下,包括多种族混合数据和高度亲缘关系数据,都表现出了良好的I型错误控制能力,确保了结果的可靠性。总而言之,LDAK-KVIK的问世为GWAS领域提供了一个强大、高效且可靠的新选择,使研究者无需再为计算资源和分析深度而烦恼,有望推动复杂性状遗传机制的进一步解析。
使用举例
为了更直观地展示其工作流程,一个典型的LDAK-KVIK分析案例如下,该流程清晰地对应了论文中描述的两步分析法。首先,需要准备标准的PLINK格式基因型文件(my_data.bed/bim/fam)、表型文件(my_pheno.phen)和协变量文件(my_covar.covar)。
第一步:构建LOCO多基因评分(PGS)
这一步是计算的核心,旨在为后续分析构建遗传背景预测模型。运行的命令如下:
./ldak6.linux --kvik-step1 kvik --bfile data --pheno pheno.pheno --covar data.covar --max-threads 2
该命令会执行计算密集型任务,包括估计SNP遗传力、构建LOCO PGS等。根据数据集的大小,这一步可能需要数小时才能完成。输出结果将保存在out/step1目录中,供第二步使用。
第二步:执行SNP关联检验
在完成第一步后,利用上一步生成的PGS作为阀值,对每个SNP进行快速的关联检验。命令如下:
./ldak6.linux --kvik-step2 kvik --bfile data --pheno phenofile --covar covfile --max-threads 4
这一步非常迅速,它会使用第一步的计算结果(通过--step1 out/step1指定)来校正群体结构和多基因背景效应,最终生成关联分析的汇总统计结果文件(out/step2.assoc),其中包含了每个SNP的P值和效应量等信息。
第三步:执行基因关联检验
./ldak6.linux --kvik-step3 kvik --bfile data --genefile genefile --max-threads 4
获得的kvik.step3.remls.all是基于基因的关联分析结果。
下载地址:https://zenodo.org/records/15747229
参考文献:Hof, J.P., Speed, D. LDAK-KVIK performs fast and powerful mixed-model association analysis of quantitative and binary phenotypes. Nat Genet (2025). https://doi.org/10.1038/s41588-025-02286-z
本篇文章来源于微信公众号:微因