
介绍snakemake之前,先上张图,这是最近自学一周snakemake写的做比较复杂的MeRIP-seq、chip-seq,ATAC-seq等的通用流程,因为想做到尽量多的通用,所以脚本逻辑复杂,坑也多,填坑花了一天。整套流程包含了:
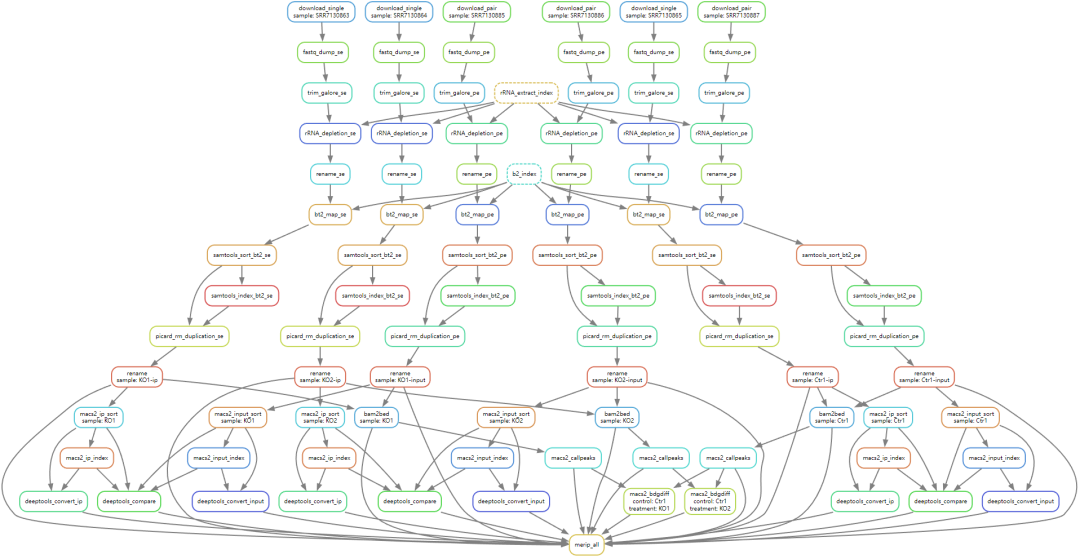
整个流程包括:
从配置文件中指定的sra连接自动下载sra测序数据 -> fastq-dump -> QC -> 去除rRNA(可选) -> 比对 -> picard去重复 -> macs2 callpeak -> macs2-bdgdiff鉴定差异 -> peak deeptools转bw。
√整套流程只需要配置好yaml文件,然后一键执行,等待结果就好;
√该流程对Ip和Input组——全是单端测序,全是双端测序以及单双端测序数据混合均可正常处理,这也是流程里逻辑比较复杂的环节之一。
后续改进计划:
√ 继续接入RNA-seq对input组进行差异表达分析,并自动生成结果报告;
√ 完成后接入到我自己的网站投入使用。
——————snakemake是啥————————
————–—snakemake的优势—————–———
√ 流程搭建简单易懂,易学习;
√ 基于python构建工作流,编程语言简单,可读性强;
√ 可重复性高。可将流程拆分成成不同的子流程,并分别编写成单个的“模块smk”,以后用于灵活拼凑不同的分析流程。比如:NGS测序数据的质控、比对、去重复分别写成单独的smk模块后,既可以用于RNA-seq数据分析的流程,也可以用于MeRIP-seq等流程中;
√ 可扩展性好:可结合conda 环境和配置文件,可以轻松实现环境的导出,扩展至其他环境下。另外,可以在rule规则中使用多种语言组合完成同一个任务,比如script既可以运行shell,python,也可以用于R,Rust等等;
√技术成熟,常规分析都能查到别人写好的流程,比如RNA-seq,Chip-seq等,网上的文档比较多、比较全。
———————-—snakemake的不足—————————
√ 官方文档杂乱繁杂,不易读懂;
√ 细节比较多,简单流程易学,但是复杂流程逻辑复杂,需要边学边用;
√ snakemake的wildcards(命名空间/通配符)相对有点难理解,有时需要顿悟,哈哈,没错,就是顿悟。
很早之前就听过snakemake了,也用过别人写的流程,但是当时没去研究过它,主要还是以前做CRIPSR实验比较多,没精力和时间。现在课题做m6A机制的,做了一堆MeRIP-seq。既然课题快整不下去了,那就把时间拿来研究一下snakemake。结果发现“相见甚早,可相识甚晚”。一个字:“棒”。
对于snakemake所需的基础,不需要太多基础,肯花时间去研究去试错就OK。主要需要稍微了解以下两点:
√ 简单的python编程语言,因为snakemake是以python3为基础去定义工作流的,换句话说,整个写的smk文件里,变量定义、路径解析、条件判断,循环执行等等全是python。但是,对于不熟悉python的也没关系,解决办法就是:简单去学习一下python常规语法,也就是简单编程就好。
推荐个学习python3的网站:https://www.runoob.com/python3/python3-tutorial.html(Python3 教程 | 菜鸟教程 (runoob.com)),这是入门教程,亲测好用,主要是免费。Get到了就记得给我一个点赞和转发。
√ 需要必备的linux基础,比如常规的命令:cd,mkdir,rm,cp,mv,awk,grep,cut,sed等等。不需要太复杂的,会用vi写简单的shell命令就好。
——————————–—下期预告———————————
√ 下期讲解如何使用conda构建snakemake的环境,以及snakemake的基础知识;
√ 构建简单的RNA-seq流程
觉得有用,不妨动动你发财的小手点个赞,然后转发一下,在下感激不尽~


扫描上方 二维码 关注我

——————————————–
祝您:月月有结果,年年有文章!
本篇文章来源于微信公众号:微因