更新亮点都有啥呢?大概是几个命令和以前不一样了。以下是发布详细信息:
QIIME 2 2020.6 版本现已发布!感谢所有参与的辛勤工作!
提醒一下,我们计划发布的 QIIME 2 计划于 2020 年 8 月发布 (QIIME 2 2020.8),但请继续关注更新。
查看QIIME 2 2020.6 文档 有关安装最新 QIIME 2 版本的详细信息,以及教程和其他资源。如果您遇到任何问题,请与QIIME 2论坛取得联系!
虚拟机镜像将在未来一周的某个时候更新!
重大更改
- q2-diversity
core_metrics_phylogenetic: 参数已重命名,以适应目前正在使用的 unifrac 新的快速实现n_jobs或者n_jobs_or_threadscore_metrics&core_metrics_phylogenetic:该短语正在替换 ,因为observed features更好地描述了用户在 QIIME 2 中使用非分类功能的不同方式(OTU 是一个feature,但feature不一定是 OTU)observed_features和observed_otuscore_metrics&core_metrics_phylogenetic:不再接受零或负整数作为参数。用户应传递给这些参数以使用所有可用的物理内核n_jobs``n_jobs_or_threads``auto
发布亮点:
- QIIME 2 框架
- 进一步开发,这将允许插件开发人员展示他们的插件操作的使用实例,一个不可知的界面方式!
这包括:
Usage API- 启用初始化数据的源跟踪
- 添加初始化数据收集的方法
- 改进了测试工具中的一些开发人员文档。
-
提高文件名的验证级别,以防止创建具有错误命名数据文件的项目。
- 进一步开发,这将允许插件开发人员展示他们的插件操作的使用实例,一个不可知的界面方式!
-
文档
- 向我们展示了如何在Moving Pictures教程中的Artifact
peek - 将一个部分添加到“导入教程“中,用于序列中具有条形码的multiplexed fastq文件。
- 阐明了
qiime tools view和 q2view 的行为 - 为使用Windows的Linux 子系统添加了用户的本机安装说明
- 添加了一些粗糙的指南,用于选择最适合您的计算需求的安装方法
- 在 Atacama 教程中添加了子采样和样本筛选工具的示例
q2-demux - 更改了建议的措辞,将 vega 编辑器用于 q2 纵向变化图以反映当前实现
- 几个小错别字被修正
- 彻底改变了用户文档托管基础结构。
- 向我们展示了如何在Moving Pictures教程中的Artifact
- 数据资源
- q2cli
- 修复了一条错误消息: 一个小拼写错误可能会导致令人惊讶的混乱!
- 合并在使用 API CLI 驱动程序的第一次迭代中。对于开发人员来说,这应该被视为”预发布”,直到使用 API 和使用示例更加成熟。敬请期待在未来版本中在此领域执行更多操作!
- QIIME 2 工作室 2
- 修复了尝试导入数据时出现的错误!
- q2 功能分类器
- 说明了我们为什么我们没有blast分类器的多线程选项
- 在
extract-reads操作中添加了一个trim-right选项,允许在 3′ 端修剪模拟读取。这对于 (例如,模拟来自 q2-dada2 的读取的修整选项)非常有用,以生成最佳修剪的参考序列以进行分类器训练,如此处所述, - 改进了
extract-reads的文档! - 使用 scikit -learn 0.23.1 训练新的功能分类器
- Q2-phylogeny
- 添加了一种新方法,
robinson-foulds用于比较两个进化树!
- 添加了一种新方法,
- q2-cutadapt
- 添加了对混合方向的间向测序multiplex reads的支持
- q2-composition
- 修复了
ancom一个错误!可视化有 (意外) 试图除以零的倾向。 - 删除了指向 ancom 绘图上的 vega 编辑器的链接, 等待 vega 5 的情节返工。
- 修复了
- Q2-feature-table
- 向
merge方法添加了一些使用示例! - 添加了在表中重新命名要素或示例的新方法
- 修复了导致
summarize可视化效果有时显示扭曲的列宽度的错误 - 删除了对汇总图上的 vega 编辑器的链接, 等待 vega 5 的情节返工
- 向
- Q2-alignment
- 添加了一个新操作 ,
mafft-add允许将未比对的序列添加到现有对齐方式
- 添加了一个新操作 ,
- Q2-quality-filter
- 合并
q-score和q-score-joined到相同的方法 (q-score),并弃用q-score-joined。此方法将在 QIIME 2 的 2020 秋季版本中删除。
- 合并
- Q2-quality-control
- 添加了一种新方法
filter_reads,该新方法支持通过SampleData[SequencesWithQuality]和SampleData[PairedEndSequencesWithQuality]比对到引用数据库来筛选和工件(也称为 FASTQ 序列数据)。你怎么能用这个?在去噪或重复之前筛选人类reads和其他非目标(或垃圾)序列。 - 过渡并使用
alpha_phylogenetic和alpha_rarefaction,这是以前在alpha_phylogenetic_alt和alpha_phylogenetic_alt中提供的速度和内存优化实现。 将在下一个版本中删除。
- 添加了一种新方法
- q2-diversity
- 添加了一个新的测试版多样性指标
jensenshannon! - 连接q2-diversity- lib 以执行
core-metrics和core-metrics-phylogenetic的 alpha 和 beta 多样性计算。有关相关API 更改,请参阅上面的”中断更改”。 - 修复了允许 0 方差数值列筛选器
bioenv导致错误的错误。
- 添加了一个新的测试版多样性指标
- q2-diversity-lib
- 此插件计算 alpha 和 beta 多样性度量,酌情扩展可用语义类型的数量,并提供特定于度量的引文信息。
- q2-types
- 添加了新的转换器,用于以有趣和有趣的方式处理每个样本序列
- 添加了更多比对序列转换器
- q2-vsearch
- 修复了
cluster-features-closed-reference方法中的性能错误,导致某些用例具有潜在的显著加速。 - 添加了一个很棒的新可视化工具
fastq_stats, 用于使用 vsearch 将单端或配对端序列蒸馏到 FASTQ 文件统计信息中! - 添加了所有 vsearch 方法接受短于 32nt 的序列的能力
- 设置
--fasta_width 0,在调用的 vsearch 命令上,以防止快速记录跨多行拆分
- 修复了
- q2-longitudinal
- 从波动性图中删除了指向 vega 编辑器的链接
- q2-demux
- 返工汇总可视化效果,以更好地通知用户缺少的reads
- q2-dada2
- 将伪池添加到所有 DADA2 工作流中,允许用户增加对样品中稀有变体(尤其是单体)的敏感性,只要在其他样本中独立观察这些变异。
- 更正了一个错误,其中仅包含一个功能的功能表被错误地诊断为空。
- q2-sample-classifier
- 给测试套件一些急需的 Tlc 与慷慨的重构。
- q2-emperor
- 更新到最新版本 1.0.1 1
-
修复了双图箭头在可视化并行绘图时会错误地显示的问题。
-
修复了在分类数据中部分使用连续和发散颜色图的问题。
-
修复了具有排序数值和非数值的 Bug。
-
修复了轴设置无法从 Python API 正确加载的问题。
-
更新了各种依赖项。
-
添加了选择一组示例并保存示例名称到用户剪贴板的能力。为此,用户需要按住Shift键并选择他们感兴趣的示例:
[
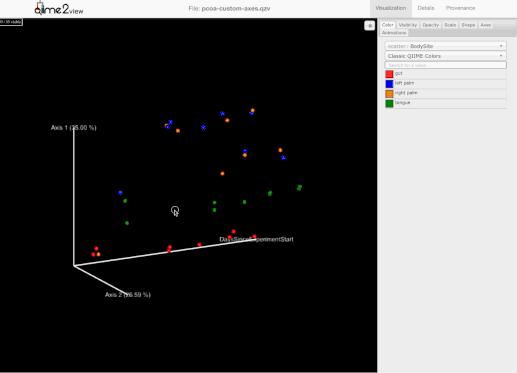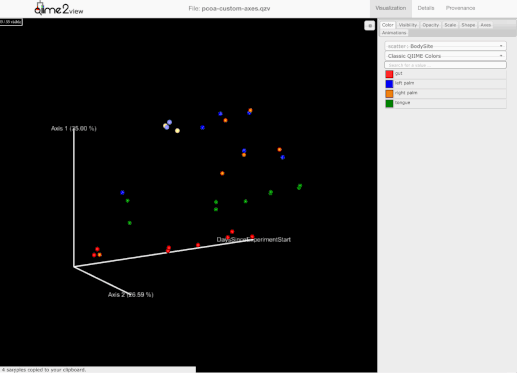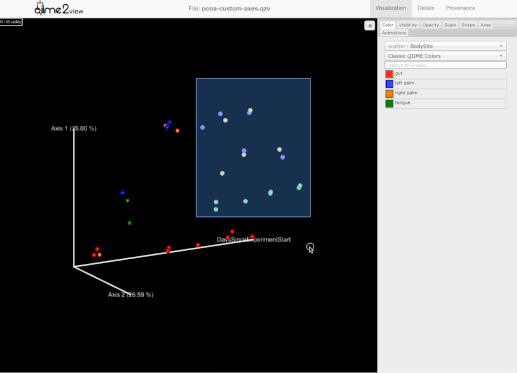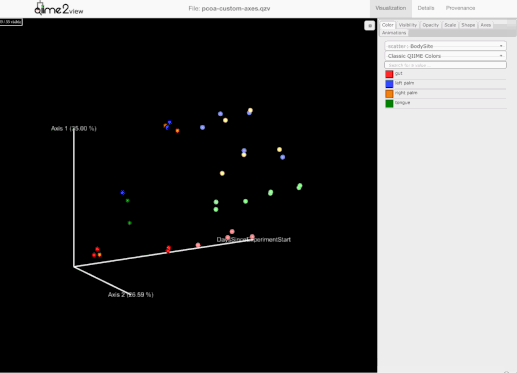
-
添加了对非混合混合方向单索引读取的支持。
快乐QIIMING!