最近有朋友和我交流纳米孔16S测序数据的分析,发现真的没有从头完成过一次这方面的数据分析,然后发现这方面的资料也比较少,于是学习一下,和大家分享。坦白说,牛津纳米孔测序技术在16S多样性研究方面还是有些不足的,只能说勉强够用,主要应用场景是在一些现场快速检测方面,主要是病原菌这种。但是,相信随着测序准确度的提高和分析软件的改进,相信它的应用会越来越多。感谢互联网的便利和分享精神,今天的我们可以方便地获得测序的原始数据,并可以自由进行分析。
1.软件和数据准备
处理牛津纳米孔的测序数据,首先当然要安装相关的专用软件了,基本上原厂出的,原厂品质,放心!还有就是minimap2和yacrd,用于去嵌合。数据来自一篇文章,文件不大,直接下载或者ascp下载均可。
#这个流程可参考https://github.com/tetedange13/stage_M2BI_scripts
#软件准备
#首先是NanoPlot,用于检测测序质量,直接pip安装了,加速的话可使用清华源
pip install NanoPlot
#去接头的Porechop
git clone https://github.com/rrwick/Porechop.git
cd Porechop
python3 setup.py install
porechop -h
#质控的NanoFilt,pip或bioconda安装
pip install nanofilt
#minimap2,编译或者下载预编译的二进制文件均可,这里编译
git clone https://github.com/lh3/minimap2
cd minimap2 && make
#yacrd,conda安装
conda install yacrd
#获取数据,我是ascp下载
/Volumes/10.11/Users/zd200572/Applications/Aspera\ Connect.app/Contents/Resources/ascp -i ~/asperaweb_id_dsa.openssh -Tr -Q -l 100M -P33001 -L- fasp-g1k@fasp.1000genomes.ebi.ac.uk:vol1/fastq/ERR277/007/ERR2778177/ERR2778177.fastq.gz .
#比对工具last,一款来自日本的比对软件,需要编译
wget http://last.cbrc.jp/last-1060.zip
unzip last-1060.zip
cd last-1060
make
#blast diamond seqkit
conda install -c bioconda diamond blast seqkit
#如果wget或者浏览器下载地址是wget http://ftp.ebi.ac.uk/vol1/fastq/ERR277/007/ERR2778177/ERR2778177.fastq.gz
#ncbi的16S数据库,由于出国网络差,下了好久,一定要确保数据下载完整,注意下载时勾选gi号,以备后面有用
#https://www.ncbi.nlm.nih.gov/nuccore?term=33175%5BBioProject%5D
#这里我把这个文件存在了网盘,公众号内回复“16S”即可获得下载地址
2.数据预处理
基本上是质控的过程,先看下测序质量,当然纳米孔的质量还是有点低的,特别是手上下载的数据是低版本的测序芯处R9.4,未来的R10可以通过两个纳米孔串联提高到95%。接着就是去除测序的接头,获得真正的测序序列,是不是引物也应该切除?但是估计数据库里的序列也应该不包含引物,所以估计引物影响不大。然后,进行过滤,除去明显不符合要求的序列。
#查看质量,fastq最常用,这里用官方的试试
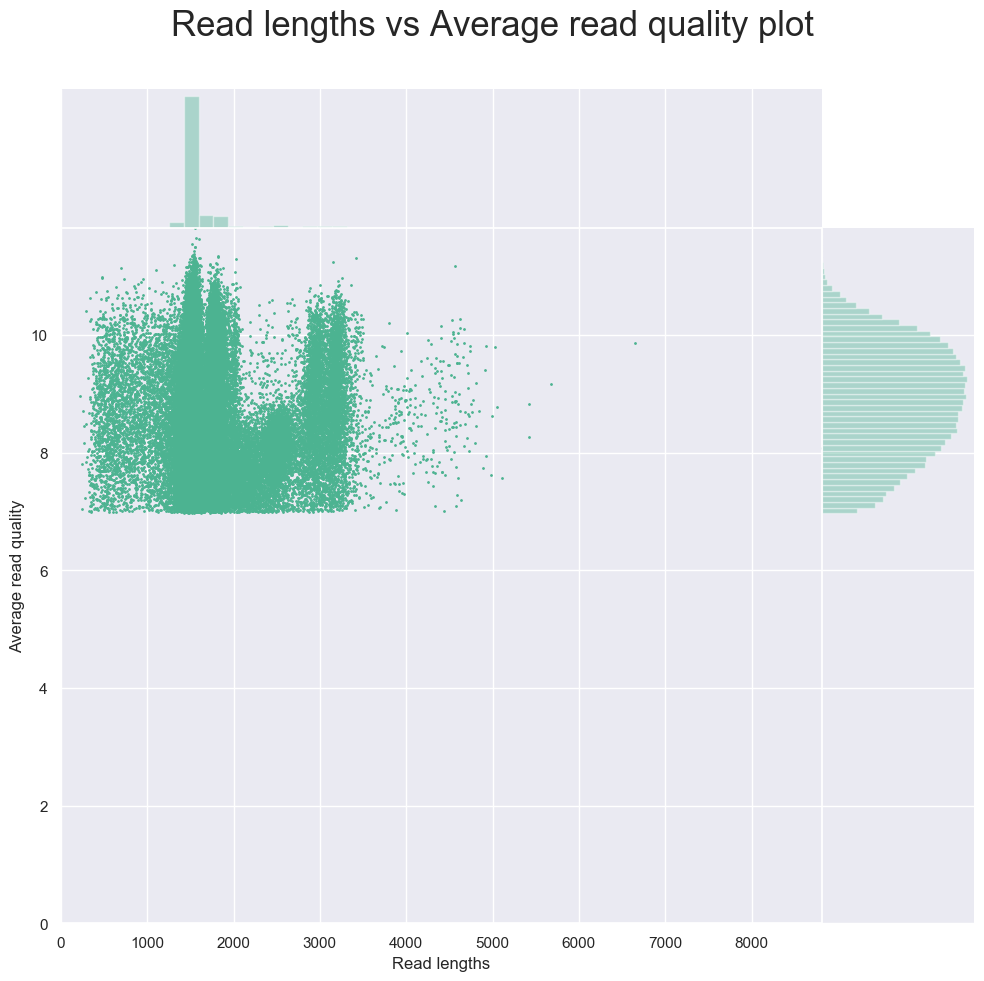
NanoPlot -t 4 --fastq ERR2778177.fastq.gz --plots hex dot
结果依然是类似于fastq质控报告的一个函数,不过统计指标少了几个,有两个把reads长度和质量分布放在一起的图不错。

#去接头,4线程,这一步耗时以小时计,对于我的五年前的双核心四线程笔记本
porechop -t 4 -i input.fastq -o trimmed.fastq
# 质控,质量值为9,长度200过滤,虽然有点低,这有两个重定向
NanoFilt -q 9 -l 200 < trimmed.fastq > cleaned.fastq
#去嵌合,先minimap2自全局比对,发现嵌合,然后yacrd去除
minimap2/minimap2 -x ava-ont -g 500 cleaned.fastq cleaned.fastq > overlap.paf
yacrd -i overlap.paf -o report.yacrd -c 4 -n 0.4 scrubb -i cleaned.fastq -o reads.scrubb.fq
#卡在这时间最长,主要几个文件要准备,需要整理的已经放在文件夹,其余的可以ncbi下载,较大,没放,或者直接用我建好的。
#看下文件的保留情况,大概去除了一半以上的数据
tree -h | grep q
├── [237M] ERR2778177.fastq.gz
├── [224M] cleaned.fastq
├── [200M] reads.scrubb.fq
└── [476M] trimmed.fastq
3. 比对
1)选项1,last比对
关于纳米孔16S的数据分析,之前翻译的那篇综述总结了大概有两种,一种是和之前的16S数据分析一样聚类ASV/OTU,但是由于90%左右的准确度,看有用85%的准确度聚类的。。。另一种就是,不聚类,直接进行比对,也就是我们这次学习的blast等比对工具的方法,根据综述作者的观点,这几种工具由于不是专门为纳米孔测序数据设计,不能比较好的完成物种注释的任务(不够准确),作者推荐的是minimap2和centrifuge这两个软件。但是,有很大一部分文章是以这几个工具进行数据分析的,我们也做一下,最后进行一个比较吧!
# 构建参考数据库
last-1060/src/lastdb -uYASS -R01 microbialdb ncbi-16S.fasta
# 比对https://gigascience.biomedcentral.com/articles/10.1186/s13742-016-0111-z#Sec12
last-1060/src/lastal -P 4 -q 1 -b 1 -Q 0 -a 1 -e 45 microbialdb cleaned.fastq > aligned.maf
# 转换成BLAST格式
last-1060/scripts/maf-convert blasttab aligned.maf > aligned.txt
2)blast/diamond比对
这个过程参考自公众号基因学苑的nanopore数据分析培训班讲义。
#w使用seqkit先转fasta
seqkit fq2fa reads.scrubb.fq > query.fasta
#建立索引,如果已经建好了,可以跳过此步骤
makeblastdb -in ncbi-16S.fasta -dbtype nucl -parse_seqids -out 16S
blastn -query query.fasta -db 16S -out blastn.out - outfmt 0 -evalue 1e-5
#利用 diamond 提高比对速度,注意 diamond 的索引与 blast 不同,需要重新建立 diamond 比对 的索引
diamond makedb --in ncbi-16S.fasta -d 16s
diamond blastx -d 16s -q query.fasta -o output.blast -p 4
4.结果处理,获得物种丰度表
由于自己水平还不够写脚本从比对结果中获得物种注释,脚本基本上来自台湾的那个同仁的。确切的说应该还是对比对结果的数据结构不够熟悉,争取后面多熟悉些。为了实现使用他的脚本,对数据库的信息进行了一些小的提取操作,用我暂时用得比较顺手的python完成的。我已经把处理好的数据上传了百度云,直接使用的话可以略过。
————–我是分界线的开始———–
cat ncbi-16S.fasta | grep ">" > conv.txt #先获得序列名信息备用
dic_fa = {}
i = 0
with open('conv.txt') as f:
for line in f:
i += 1
if line.strip() != '':
ac_n = line.strip().split('|')[3]
gi_n = line.strip().split('|')[1]
dic_fa[ac_n] = ''
dic_fa[ac_n] = [gi_n, line.strip()]
print(i)
j = 0
fout = open('info.txt', 'w')
with open('sequence.gb') as f1:
for line in f1:
if line[:7] == "VERSION":
seq_name = line.strip().split(' ')[1].split(' ')[0]
if '/db_xref="taxon:' in line:
tax_id = line.strip().split('/db_xref="taxon:')[1].split('"')[0]
if seq_name in dic_fa.keys():
fout.write(dic_fa[seq_name][1] + '\t' + tax_id + '\n') #+ seq_name + '\n')
seq_name = ''
tax_id = ''
j += 1
print(j)
fout.close()
————–我是分界线的结束———–
# 加载包,第一次看到这样的方式
suppressMessages(library(data.table))
suppressMessages(library(tidyverse))
# 设置文件位置
id_mapping_file <- "/Volumes/Data/Biodata/Nanopore-16S/info.txt"
blastOutputs <- "/Volumes/Data/Biodata/Nanopore-16S/aligned.txt"
#导入输入文件
id_mapping <- fread(id_mapping_file, header = FALSE, sep = "\t", stringsAsFactors = FALSE)
setkey(id_mapping, V1)
blastoutput <- fread(blastOutputs, header = FALSE, sep = "\t", stringsAsFactors = FALSE)
blastoutput_filtered <- blastoutput %>%
dplyr::filter(V3>=90 & V4>=700) %>%
group_by(V1) %>%
dplyr::filter(V3 == max(V3)) %>%
dplyr::filter(V12 == max(V12))
allreadsnum <- length(unique(blastoutput$V1))
readStats <- table(blastoutput_filtered$V2)
refseqIds <- names(readStats)
df <- data.frame(RefseqID = refseqIds,
Lineage = NA,
ReadsNum = NA,
OrgName = NA,
ReadsPerc = NA)
#循环获得信息
for (i in 1:length(refseqIds)) {
refseqid <- refseqIds[i]
tmp <- id_mapping[.(refseqid)]
lineage <- tmp$V3
orgname <- tmp$V4
readnum <- readStats[refseqid]
perc <- readnum*100/allreadsnum
df$Lineage[i] <- lineage
df$ReadsNum[i] <- readnum
df$ReadsPerc[i] <- perc
df$OrgName[i] <- orgname
}
taxonomy <- paste(df$Lineage, df$OrgName, sep="; ")
output_df <- data.frame(Taxonomy = taxonomy, ReadsNumber = df$ReadsNum)
output_df <- aggregate(output_df$ReadsNumber,
by = list(Taxonomy=output_df$Taxonomy),
FUN = sum)
colnames(output_df)[2] <- "ReadsNumber"
ReadPercentage <- output_df$ReadsNumber*100/allreadsnum
output_df <- cbind(output_df, ReadPercentage)
output_df <- output_df[order(output_df$ReadPercentage, decreasing = TRUE),]
#output <- paste0("tax_", sub("\\.\\/Alignment\\/", "", file))
write.table(output_df, 'out.txt', sep = "\t", row.names = FALSE, quote = FALSE)
最后,如果顺利,就是结果了:
| Taxonomy | ReadsNumber | ReadPercentage |
|---|---|---|
| Trichocoleus desertorum strain ATA4-8-CV2 | 1046 | 41.85674270 |
| Neosynechococcus sphagnicola strain sy1 | 248 | 9.92396959 |
| Tychonema bourrellyi strain CCAP 1459/11B | 110 | 4.40176070 |
| Kastovskya adunca strain ATA6-11-RM4 | 104 | 4.16166467 |
| Okeania plumata strain FK12-27 | 74 | 2.96118447 |
| Loriellopsis cavernicola strain LF-B5 | 53 | 2.12084834 |
| Cephalothrix komarekiana CCIBt 3277 | 42 | 1.68067227 |
| Caedimonas varicaedens strain 221 | 41 | 1.64065626 |
| Aliterella antarctica strain CENA408 | 33 | 1.32052821 |
| Limnoraphis robusta strain CCALA 966 | 31 | 1.24049620 |