最近看到生信技能树的一篇推文在介绍nf-core这个流程管理工具,发现官方有qiime2的流程,学习一下,顺便探索一下中间的坑。关于nf-core,这篇推文已经介绍的够多了,我这里主要学习它的搭建和使用。
一、环境搭建
首先,先进行环境搭建工作,这是必修课和基础,没有环境,什么也做不了。理解下来,nf-core可以使用三种方式进行环境准备,本地安装,conda或者docker,一般来说,对新手最友好的当属conda了,除了有的软件清华源镜像里没有,会速度极慢,容易失败,可能环境准备要放许久,如果数据不大的话,建议选用一台物理地址在香港等地的小云服务器解决,软件安装节省很多很多时间。
#下载conda,境内推荐清华源
#https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda3-latest-Linux-x86_64.sh
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
#按照提示安装
bash Miniconda3-latest-Linux-x86_64.sh
#如果选择不初始化,激活环境
source miniconda3/bin/activate
#下载流程所需要的环境配置文件
wget https://github.com/nf-core/ampliseq/raw/master/environment.yml
#创建流程所需要的环境
conda env create -n ampliseq --file environment.yml
#激活环境
conda activate ampliseq
#安装nextflow
conda install -c bioconda nextflow -y
二、配置和运行
配置主要是参考github上这个流程的参数说明,主要是控制16S的扩增引物,电脑的最大CPU核心数和RAM,序列质控trim的长度,先fastqc确定一下。
#配置
cd test
#把数据放在工作目录,这里省略
#配置好sample-metadata.txt样本信息表,下载已经训练好的分类参考
#版本需要对应,这里是2019.10
wget https://data.qiime2.org/2019.10/common/gg-13-8-99-nb-classifier.qza
#然后运行流程,这里我开了一个虚拟机,双核4g
#因为已经切到建好的环境了,就不再加上-profile conda参数了,否则又要新建一个一样的环境
nextflow run nf-core/ampliseq --reads "Dong-16S" \
--FW_primer TACGGRAGGCAGCAG \
--RV_primer AGGGTATCTAATCCT \
--metadata "sample-metadata.txt" \
--untilQ2import \
--extension "/*R{1,2}.fastq" \
--trunclenf 280 \
--trunclenr 250 \
--max_memory '3.GB' \
--max_cpus 2 \
--onlyDenoising
然后,就得到了输出结果:
给我的感觉是,一个成熟的流程构建者由于对数据处理有丰富的经验,可以充分地利用计算机的硬件最大潜能,实现最短的时间完成最大的任务量,这对于生产环境是用及其重要的,科研环境一般可能不会有这种问题,科研最需要的是画图,和能说明问题的结论以及故事。它充分地合理安排了各个任务,可以步骤交替运行,但基本上没有限速步骤,这是值得学习和使用的地方。
Launching `nf-core/ampliseq` [reverent_goldstine] - revision: cd23988d88 [master]
[2m----------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/ampliseq v1.1.2
----------------------------------------------------
Pipeline Name : nf-core/ampliseq
Pipeline Release : master
Run Name : reverent_goldstine
Reads : Dong-16S
Data Type : Paired-End
Max Resources : 3.GB memory, 2 cpus, 10d time per job
Output dir : ./results
Launch dir : /root/test_project
Working dir : /root/test_project/work
Script dir : /root/.nextflow/assets/nf-core/ampliseq
User : root
Config Profile : standard
------------------------------------------------------
executor > local (1)
[- ] process > get_software_versions [ 0%] 0 of 1
[34/c556b9] process > fastqc [ 0%] 0 of 6
[- ] process > trimming [ 0%] 0 of 6
[- ] process > multiqc -
[- ] process > qiime_import -
[- ] process > qiime_demux_visualize -
......
[d8/db2448] process > get_software_versions [100%] 1 of 1 ✔
[2e/88b642] process > fastqc [100%] 6 of 6 ✔
[cf/4605c1] process > trimming [100%] 6 of 6 ✔
[57/54f4cc] process > multiqc [100%] 1 of 1 ✔
[9e/b298db] process > qiime_import [100%] 1 of 1 ✔
[8b/60b53e] process > qiime_demux_visualize [100%] 1 of 1 ✔
[08/804cd9] process > dada_trunc_parameter [100%] 1 of 1 ✔
[5c/532077] process > dada_single [100%] 1 of 1 ✔
[25/c6cdaa] process > classifier [100%] 1 of 1 ✔
[2b/ef2011] process > filter_taxa [100%] 1 of 1 ✔
[5f/cf7385] process > export_filtered_dada_output [100%] 1 of 1 ✔
[29/caccdc] process > report_filter_stats [100%] 1 of 1 ✔[63/2018d3] process > RelativeAbundanceASV [100%] 1 of 1 ✔
[35/521394] process > RelativeAbundanceReducedTaxa [100%] 1 of 1 ✔[e3/06d354] process > barplot [100%] 1 of 1 ✔
[cb/b1321a] process > tree [100%] 1 of 1 ✔[62/9aab63] process > alpha_rarefaction [100%] 1 of 1 ✔
[0b/caf4ae] process > combinetable [100%] 1 of 1 ✔[6d/00777a] process > diversity_core [100%] 1 of 1 ✔
[13/37d1de] process > metadata_category_all [100%] 1 of 1 ✔
[81/ac4e94] process > metadata_category_pairwise [100%] 1 of 1 ✔
[0e/77008d] process > alpha_diversity [100%] 4 of 4, failed: 4 ✔
[- ] process > beta_diversity -
[52/e9e491] process > beta_diversity_ordination [100%] 4 of 4 ✔
[02/1173eb] process > prepare_ancom [100%] 1 of 1 ✔
[93/fec20e] process > ancom_tax [100%] 5 of 5 ✔
[ca/f279b2] process > ancom_asv [100%] 1 of 1 ✔
[ae/e29fd4] process > output_documentation [100%] 1 of 1 ✔
[0;35mWarning, pipeline completed, but with errored process(es)
[0;31mNumber of ignored errored process(es) : 4
[0;32mNumber of successfully ran process(es) : 43
[0;35m[nf-core/ampliseq] Pipeline completed successfully
[a9/5c27da] NOTE: Process `alpha_diversity (evenness_vector)` terminated with an errorexit status (1) -- Error is ignored
WARN: To render the execution DAG in the required format it is required to install Graphviz -- See http://www.graphviz.org for more info.
Completed at: 08-Apr-2020 06:44:42
Duration : 9m 24s
CPU hours : 0.2 (2.4% failed)
Succeeded : 43
Ignored : 4
Failed : 4
#运行时间9分钟左右,已经超级高效了,我手动做的话会到法小时吧。因为手上数据有些质量问题,处理过程中有报错
三、结果欣赏
来看看这个结果怎样,因为结果做的很漂亮,所以用上了欣赏这个词。基本上相当于一个公司的数据分析报告的感觉,我觉得如果再加上一个网页端,人人都可以云生信做微生物数据分析了。毕竟,16S数据分析也不需要多强大的电脑,自己的笔记本就可以搞定。专注于具体的参数,而不需要考虑每一个命令,这就是未来呀。从运行过程来看,作者还使用了一些R脚本完成了许多图形的绘制,以及部分文件的操作。
#安装tree,查看文件目录树
sudo apt install tree
tree
#以下是输出
├── Documentation
├── MultiQC
├── abundance_table
├── alpha-diversity
├── alpha-rarefaction
├── ancom
├── barplot
├── beta-diversity
├── demux
├── fastQC
├── phylogenetic_tree
├── pipeline_info
├── rel_abundance_tables
├── representative_sequences
├── taxonomy
└── trimmed
1.提供了一个帮助文档,方便理解以上各个文件的信息。

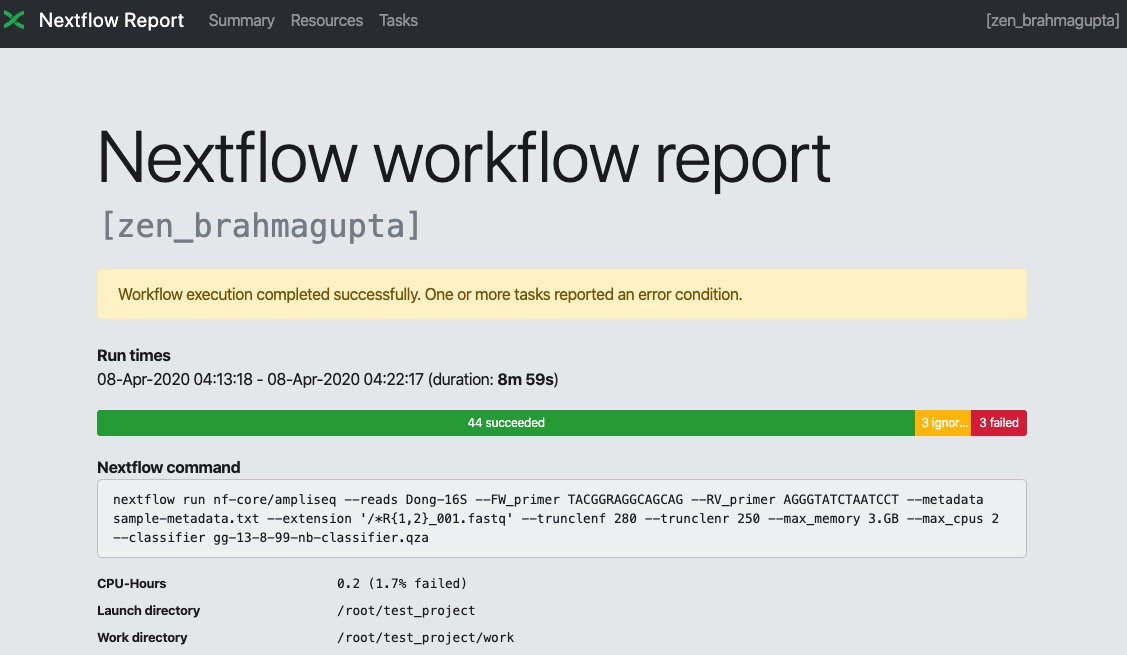
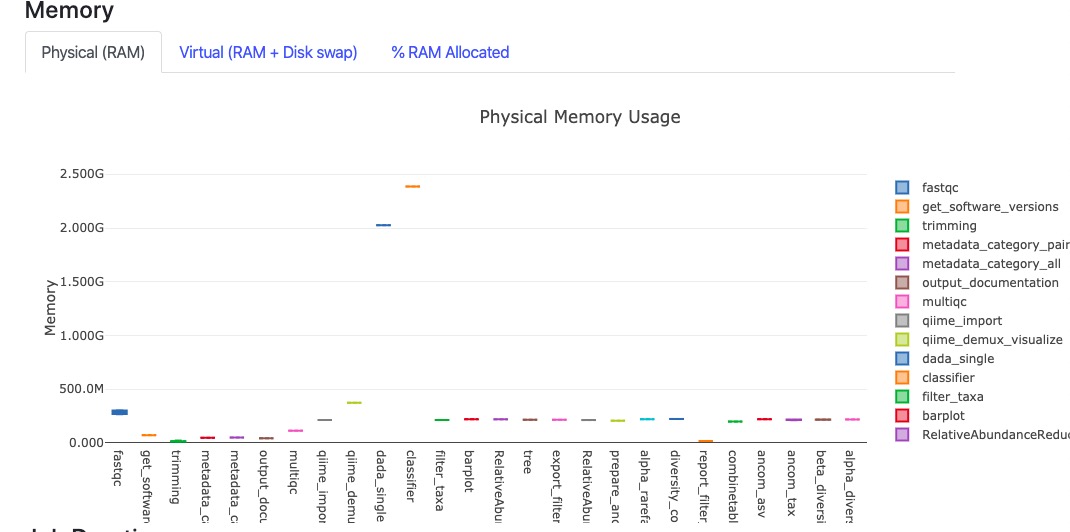
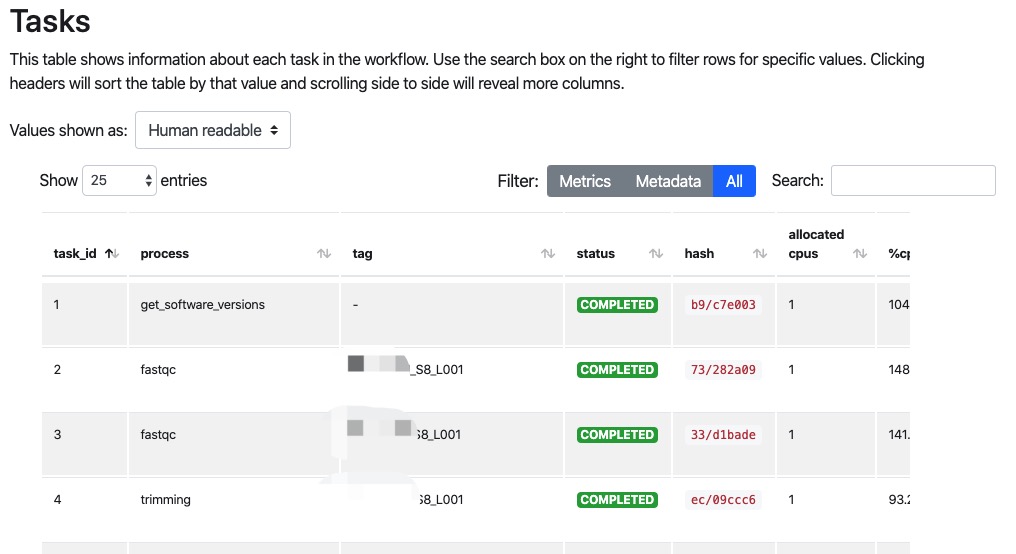
2.然后是结果汇总,是流程的运行概览信息,CPU,内存使用情况和运行时间,以及各个任务的详细信息,包括脚本命令等。



3.关于结果,流程是把qiime2的qzv格式做了解压处理,这样方便直接用网页打开而不需要view.qiime2.cn这个网站。而且对文件进行了重命名,方便进行查阅。和qiime2的输出结果是一样的,这里就不放了。