这项工作已获得知识共享署名-相同方式共享4.0国际协议的许可。这意味着您可以复制,共享和修改作品,只要结果以相同的许可证分发即可。
本教程由Mobolaji Adeolu(adeolum@mcmaster.ca),John Parkinson(john.parkinson@utoronto.ca)和Xuejian Xiong(xuejian@sickkids.ca)制作。
注意,这个教程的软件运行环境为linux,没有相关环境需要使用docker或者虚拟机,而且,经过测试,python版本要求为2.7, biopython=1.67,在不停报错的教训中得到的结论。
总览
本教程将带您完成处理宏转录组数据的流程。实验室开发的reads包括以下各个步骤:
- 除去在文库制备和测序步骤中添加的衔接子序列,并修剪低质量的碱基和测序读数。
- 删除重复的reads以减少以下步骤的处理时间。
- 除去载体污染(来自克隆载体,刺突和引物的读数)。
- 除去宿主读物(如果要研究其中存在宿主的微生物组)。
- 尽管使用了rRNA去除试剂盒,但仍要删除通常主导转录组数据集的大量rRNA序列。
- 将重复的reads(在步骤2中删除)添加回数据集,以提高程序集的质量。
- 将reads分类到已知的分类组,并可视化数据集的分类组成。
- 将读段组装到重叠群中以提高注释质量。
- 注释reads已知基因。
- 将已鉴定的基因映射到swiss-prot数据库中以鉴定酶功能
- 生成与每个基因相关的标准化表达值。
- 使用KEGG代谢途径作为Cytoscape的支架,可视化结果。
整个元转录组学流程包括现有的生物信息学工具和一系列处理文件格式转换和输出解析的Python脚本。我们将通过以下步骤来说明流程的复杂性以及基础工具和脚本。
一直在开发新的,更快和/或更准确的工具,值得牢记的是,随着这些流程被社区采纳为标准,任何流程都需要灵活地整合这些工具。例如,在过去的两年中,我们的实验室已经从cross_match过渡到Trimmomatic,从BLAST过渡到DIAMOND。注意:本研讨会旨在与DIAMOND v0.826一起使用。较新版本的DIAMOND将与我们在此练习中制作的预编译数据库文件不兼容。
为了说明该过程,我们将使用从小鼠结肠内容产生的序列reads。这些是150 bp单端reads。也可以使用成对末端的reads,并且通常是首选的,因为当reads对中有足够的重叠以提高有效平均reads长度时,它们可以提高注释质量。使用成对末端数据需要一个额外的数据处理步骤(合并重叠的reads),从而在数据处理过程中生成更多文件(用于合并/单reads,正向reads和反向reads的文件),但是成对末端reads的结构数据类似于此处描述的reads,并且可以轻松进行调整。
本教程将带您逐步处理100000个reads的一部分,而不是使用整个2500万个reads的整个过程(在桌面上可能要花费几天的时间)。
开场
工作目录
#创建一个新目录,该目录将存储在本实验中创建的所有文件。
mkdir -p ~/metatranscriptomics
cd ~/metatranscriptomics
Python脚本
我们已经编写了许多脚本来从您将要使用的工具中提取和分析数据。下载用于元转录组学研讨会的软件包,并解压缩我们的python脚本。
#原文采用wget,这里为了加速,采用多线程的axel
axel https://github.com/ParkinsonLab/2017-Microbiome-Workshop/releases/download/Extra/precomputed_files.tar.gz
wget https://jiawen.zd200572.com/hla/precomputed_files.tar.gz --no-check-certificate
wget https://github.com/ParkinsonLab/Metatranscriptome-Workshop/archive/EC.zip --no-check-certificate
unzip EC.zip
tar -zxvf precomputed_files.tar.gz *.py --wildcards
输入文件
我们的数据集包含从小鼠结肠内容物产生的150 bp单端Illumina读数。要检查其内容:
tar -xvf precomputed_files.tar.gz mouse1.fastq
less mouse1.fastq
注意事项:
输入q以退出less。
使用FastQC检查reads质量
FastQC报告在HTML文件中生成mouse1_fastqc.html。您还将找到一个zip文件,其中包含用于生成报告的数据文件。
source ~/Miniconda/bin/activate
conda create -n metatranscripts python=2.7
conda activate metatranscripts
conda install -y bwa SPAdes fastqc biopython=1.67 diamond=0.826 blat blast samtools bwa
fastqc mouse1.fastq -t 4 #只有一个文件,所以4线程是可选的
FastQC报告在HTML文件中生成mouse1_fastqc.html。您还将找到一个zip文件,其中包含用于生成报告的数据文件。
要打开HTML报告文件,请使用以下命令, 然后您用浏览器可以浏览mouse1_fastqc.html并查找以下信息:
基本统计信息:小鼠RNA序列数据的基本信息,例如reads总数,reads长度,GC含量。
每碱基序列质量:每个位置上所有碱基的质量值范围的概述。
每碱基序列含量:显示跨序列长度的核苷酸偏差的图。
适配器内容:提供有关序列样品中适配器污染程度的信息。
处理reads
步骤1.移除适配器序列并修剪低质量序列。
Trimmomatic可以快速识别和修剪适配器序列,以及识别和删除低质量序列数据
注意事项:
”’
Automatically using 4 threads
Using Long Clipping Sequence: ‘AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTA’
Using Long Clipping Sequence: ‘AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC’
ILLUMINACLIP: Using 0 prefix pairs, 2 forward/reverse sequences, 0 forward only sequences, 0 reverse only sequences
Quality encoding detected as phred33
Input Reads: 100000 Surviving: 94415 (94.42%) Dropped: 5585 (5.58%)
TrimmomaticSE: Completed successfully
”’
ln -s /usr/local/prg/Trimmomatic-0.36/adapters/TruSeq3-SE.fa Adapters用于创建指向Trimmomatic提供的单端适配器序列文件的符号链接,该文件适用于HiSeq和MiSeq机器生成的序列。但是,如果可能,应使用您自己的测序项目中的已知适配器文件替换此文件。
命令行参数是:
SE:输入数据是单端reads。
ILLUMINACLIP:Adapters:2:30:10:卸下适配器。
LEADING:3:如果reads的质量得分低于3,则在reads开始时将其作为基础。
TRAILING:3:如果它们的质量得分低于3,则在reads结束时修剪基数。
SLIDINGWINDOW:4:15:使用大小为4的窗口进行扫描,以reads局部质量低于15的读数,如果发现则进行修剪。
MINLEN:50:删除长度小于50的序列。
问题1:删除了多少个低质量序列?
ln -s ~/Miniconda/envs/tera/share/trimmomatic-*/adapters/TruSeq3-SE.fa Adapters
#ln -s /usr/local/prg/Trimmomatic-0.36/adapters/TruSeq3-SE.fa Adapters
Trimmomatic SE mouse1.fastq mouse1_trim.fastq ILLUMINACLIP:Adapters:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50
#运行过程的输出
Automatically using 4 threads
Using Long Clipping Sequence: 'AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTA'
Using Long Clipping Sequence: 'AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC'
ILLUMINACLIP: Using 0 prefix pairs, 2 forward/reverse sequences, 0 forward only sequences, 0 reverse only sequences
Quality encoding detected as phred33
Input Reads: 100000 Surviving: 94415 (94.42%) Dropped: 5585 (5.58%)
TrimmomaticSE: Completed successfully
ln -s /usr/local/prg/Trimmomatic-0.36/adapters/TruSeq3-SE.fa Adapters用于创建指向Trimmomatic提供的单端适配器序列文件的符号链接,该文件适用于HiSeq和MiSeq机器生成的序列。但是,如果可能,应使用您自己的测序项目中的已知适配器文件替换此文件。
命令行参数是:
SE:输入数据是单端reads。
ILLUMINACLIP:Adapters:2:30:10:卸下适配器。
LEADING:3:如果reads的质量得分低于3,则在reads开始时将其作为基础。
TRAILING:3:如果它们的质量得分低于3,则在reads结束时修剪基数。
SLIDINGWINDOW:4:15:使用大小为4的窗口进行扫描,以reads局部质量低于15的读数,如果发现则进行修剪。
MINLEN:50:删除长度小于50的序列。
问题1:删除了多少个低质量序列?
使用FastQC检查reads质量:fastqc mouse1_trim.fastq -t 4
与上一份报告进行比较,以查看以下各节中的更改:
基本统计
每碱基序列质量
可选:配对读合并
如果您使用的是配对末端数据集,我们可以识别重叠的序列读对,因此可以合并为单个序列。为此,我们使用工具VSEARCH,可以在以下网站上找到该工具:
Exmaple only, do not run!
vsearch --fastq_mergepairs mouse1_trim.fastq --reverse mouse2_trim.fastq --fastqout mouse_merged_trim.fastq --fastqout_notmerged_fwd mouse1_merged_trim.fastq --fastqout_notmerged_rev mouse2_merged_trim.fastq
注意事项:
命令行参数是:
–fastq_mergepairs 指示VSEARCH使用reads合并算法来合并重叠的成对末端reads
–reverse 指示具有3’至5’(反向)配对末端reads的文件名
–fastqout 指示输出文件包含重叠的配对末端reads
–fastqout_notmerged_fwd和–fastqout_notmerged_rev指示输出文件包含不重叠的成对末端读
如果要查看合并的reads长度的分布,可以使用fastqc检查reads属性:
fastqc mouse_merged_trim.fastq
firefox mouse_merged_trim_fastqc.html
reads质量过滤
Trimmomatic用于删除读物中的衔接子和修剪低质量碱基,它使用滑动窗口方法删除reads中低质量碱基的连续区域。但是,值得强加一个总体reads质量阈值,以确保在我们的分析中使用的所有reads均具有足够的无差错。为此,我们使用可在此网站上找到的工具VSEARCH (在处理配对末端数据时,此步骤应在reads合并步骤之后执行):
vsearch --fastq_filter mouse1_trim.fastq --fastq_maxee 2.0 --fastqout mouse1_qual.fastq
注意事项:
命令行参数是:
–fastq_filter 指示VSEARCH使用质量过滤算法删除低质量reads
–fastq_maxee 2.0预期的错误阈值。设置为1。任何质量得分表明平均预期错误数量大于1的读数都将被过滤。
–fastqout 指示输出文件包含高质量的过滤reads
使用FastQC检查reads质量:
fastqc mouse1_qual.fastq
mouse1_qual_fastqc.html与以前的报告进行比较,以查看以下各节中的更改:
基本统计
每碱基序列质量
每序列质量
问题2:每次reads序列质量曲线如何变化?
步骤2.删除重复的reads
为了大大减少识别和过滤rRNA读段所需的计算时间,我们使用可从本网站获得的软件工具CD-HIT执行去重复步骤,以删除重复的读段。
wget https://jiawen.zd200572.com/hla/cd-hit-v4.8.1-2019-0228.tar.gz --no-check-certificate
tar zxvf cd-hit-v4.8.1-2019-0228.tar.gz
cd cd-hit-v4.8.1-2019-0228/
make openmp=no
cd cd-hit-auxtools
make
cd-hit-v4.8.1-2019-0228/cd-hit-auxtools/cd-hit-dup -i mouse1_qual.fastq -o mouse1_unique.fastq
注意事项:
命令行参数是:
-i:输入的fasta或fastq文件。
-o:包含去重复序列的输出文件,其中唯一的代表序列用于表示具有多个重复的每组序列。
mouse1_unique.fastq.clstr创建第二个输出文件,该文件确切显示由去复制的文件中的每个唯一序列表示的复制序列,mouse1_unique.fastq2.clstr还创建了第三个空的输出文件,该文件仅用于配对末端reads。
问题3:您能找到多少个独特的reads内容吗?
尽管在这个小型数据集中复制的reads次数相对较少,但对于较大的数据集,此步骤可以将文件大小减少多达50-80%
步骤3.去除载体污染
为了识别和过滤来自载体,衔接子,接头和引物污染源的读数,我们使用了Burrows Wheeler序列比对器(BWA)和BLAST样比对工具(BLAT)来搜索奶牛序列数据库。作为用于识别污染性载体和衔接子序列的参考数据库,我们依赖于UniVec_Core数据集,该数据集是已知载体以及从NCBI Univec数据库派生的常见测序衔接子,接头和PCR引物的fasta文件。请首先将其下载到您的工作目录中。
axel ftp://ftp.ncbi.nih.gov/pub/UniVec/UniVec_Core
现在,我们必须使用以下命令为BWA和BLAT的这些序列生成索引:
接下来,我们可以使用BWA对读段进行对齐,并使用以下命令使用Samtools筛选出与矢量数据库对齐的所有读段:
bwa index -a bwtsw UniVec_Core
samtools faidx UniVec_Core
makeblastdb -in UniVec_Core -dbtype nucl
接下来,我们可以使用BWA对读段进行对齐,并使用以下命令使用Samtools筛选出与矢量数据库对齐的所有读段:
bwa mem -t 4 UniVec_Core mouse1_unique.fastq > mouse1_univec_bwa.sam
samtools view -bS mouse1_univec_bwa.sam > mouse1_univec_bwa.bam
samtools fastq -n -F 4 -0 mouse1_univec_bwa_contaminats.fastq mouse1_univec_bwa.bam
samtools fastq -n -f 4 -0 mouse1_univec_bwa.fastq mouse1_univec_bwa.bam
注意事项:
用于执行以下任务的命令:
bwa mem:生成与载体污染物数据库的读数比对
samtools view:将bwa的.sam输出转换为.bam,以进行以下步骤
samtools fastq:生成所有的fastq输出reads映射到向量污染物数据库(-F 4)和所有reads没有映射到向量污染物数据库(-f 4)
问题4:您能否找到映射到向量数据库的BWAreads数目?
现在,我们想对使用BLAT的读段执行其他比对,以滤除与载体污染数据库对齐的所有剩余reads。但是,BLAT仅接受fasta文件,因此我们必须将reads内容从fastq转换为fasta。可以使用VSEARCH完成。
vsearch --fastq_filter mouse1_univec_bwa.fastq --fastaout mouse1_univec_bwa.fasta
注意事项:
所使用的VSEARCH命令–fastq_filter与步骤1中用于过滤低质量reads的命令相同。但是,这里我们没有提供过滤条件,因此所有输入reads都传递到输出fasta文件。
现在,我们可以使用BLAT对载体污染数据库进行额外的比对。
blat -noHead -minIdentity=90 -minScore=65 UniVec_Core mouse1_univec_bwa.fasta -fine -q=rna -t=dna -out=blast8 mouse1_univec.blatout
注意事项:
命令行参数是:
-noHead:禁止.psl标头(因此它只是一个制表符分隔的文件)。
-minIdentity:设置最小序列同一性为90%。
-minScore:设置最低分数为65。这是匹配减去不匹配减去某种空位罚分。
-fine:用于高质量的mRNA。
-q:查询类型为RNA序列。
-t:数据库类型为DNA序列。
最后,我们可以运行一个小的python脚本来过滤BLAT不能自信地与我们的载体污染数据库中的任何序列比对的读数。
注意事项:
./1_BLAT_Filter.py mouse1_univec_bwa.fastq mouse1_univec.blatout mouse1_univec_blat.fastq mouse1_univec_blat_contaminats.fastq
注意事项:
该脚本的参数结构为: 1_BLAT_Filter.py <Input_Reads.fq>
在这里,BLAT不会识别与载体污染物数据库比对的任何其他序列。但是,我们发现BLAT通常能够找到BWA无法识别的比对,特别是在搜索由全基因组组成的数据库时。
在数百万个大型reads数据集中对BWA遗漏的矢量污染物进行了一些比对。
步骤4.删除主机reads
为了识别和过滤宿主读物(这里是小鼠来源的读物),我们使用小鼠DNA序列数据库重复上述步骤。为了我们的目的,我们使用从Ensembl下载的小鼠基因组数据库。
wget ftp://ftp.ensembl.org/pub/current_fasta/mus_musculus/cds/Mus_musculus.GRCm38.cds.all.fa.gz
gzip -d Mus_musculus.GRCm38.cds.all.fa.gz
mv Mus_musculus.GRCm38.cds.all.fa mouse_cds.fa
然后,我们重复上述步骤,为BWA和BLAT的这些序列生成索引:
bwa index -a bwtsw mouse_cds.fa
samtools faidx mouse_cds.fa
makeblastdb -in mouse_cds.fa -dbtype nucl
现在,我们使用BWA和Samtools对齐并过滤出与我们的宿主序列数据库对齐的所有reads:
bwa mem -t 4 mouse_cds.fa mouse1_univec_blat.fastq > mouse1_mouse_bwa.sam
samtools view -bS mouse1_mouse_bwa.sam > mouse1_mouse_bwa.bam
samtools fastq -n -F 4 -0 mouse1_mouse_bwa_contaminats.fastq mouse1_mouse_bwa.bam
samtools fastq -n -f 4 -0 mouse1_mouse_bwa.fastq mouse1_mouse_bwa.bam
最后,我们使用BLAT对我们的宿主序列数据库进行额外的比对。
vsearch --fastq_filter mouse1_mouse_bwa.fastq --fastaout mouse1_mouse_bwa.fasta
blat -noHead -minIdentity=90 -minScore=65 mouse_cds.fa mouse1_mouse_bwa.fasta -fine -q=rna -t=dna -out=blast8 mouse1_mouse.blatout
./1_BLAT_Filter.py mouse1_mouse_bwa.fastq mouse1_mouse.blatout mouse1_mouse_blat.fastq mouse1_mouse_blat_contaminats.fastq
问题5:BWA和BLAT与小鼠宿主序列数据库比对了多少次?
可选:在您自己的未来分析中,您可以选择使用向量污染数据库和宿主序列数据库的组合来同时完成步骤3和4cat UniVec_Core mouse_cds.fa > contaminants.fa。但是,一起执行这些步骤使您很难分辨您的读物中有多少是专门来自宿主生物的。
步骤5.删除大量rRNA序列
rRNA基因倾向于在所有样品中高度表达,因此必须进行筛选,以避免组装和注释步骤的下游下游处理时间过长。您可以在此步骤中使用序列相似性工具(例如BWA或BLAST),但是我们发现[Infernal](http://infernal.janelia.org/)速度较慢,但 它更敏感,因为它依赖于协方差模型数据库( CMs)描述基于Rfam数据库的rRNA序列图。由于对CM的依赖,Infernal在单个内核上进行约100,000次reads最多可能需要4个小时。因此,我们将跳过此步骤,并使用mouse1_rRNA.infernalouttar文件中的预计算文件precomputed_files.tar.gz。
tar -xzf precomputed_files.tar.gz mouse1_rRNA.infernalout
注意事项:
下面给出了用于使用地狱生成此输出的命令:
命令行参数是:
-o:地狱输出日志文件。
–tblout:简单的表格输出文件。
–noali:从主输出中省略对齐部分。这样可以大大减少输出量。
–anytrunc:放宽截断对齐的阈值
–rfam:使用针对大型数据库设计的严格过滤策略。这将加快搜索速度,但可能会降低灵敏度。
-E:报告E值为0.001的靶序列。
从此输出文件中,我们需要使用脚本来过滤rRNAreads:
vsearch --fastq_filter mouse1_mouse_blat.fastq --fastaout mouse1_mouse_blat.fasta
cmsearch -o mouse1_rRNA.log --tblout mouse1_rRNA.infernalout --anytrunc --rfam -E 0.001 Rfam.cm mouse1_mouse_blat.fasta
命令行参数是:
-o:地狱输出日志文件。
–tblout:简单的表格输出文件。
–noali:从主输出中省略对齐部分。这样可以大大减少输出量。
–anytrunc:放宽截断对齐的阈值
–rfam:使用针对大型数据库设计的严格过滤策略。这将加快搜索速度,但可能会降低灵敏度。
-E:报告E值为0.001的靶序列。
从此输出文件中,我们需要使用脚本来过滤rRNAreads:
./2_Infernal_Filter.py mouse1_mouse_blat.fastq mouse1_rRNA.infernalout mouse1_unique_mRNA.fastq mouse1_unique_rRNA.fastq
注意事项:
该脚本的参数结构为: 2_Infernal_Filter.py <Input_Reads.fq> <Infernal_Output_File> <mRNA_Reads_Output> <rRNA_Reads_Output>
在这里,我们只删除了数千个读段,而不是映射到rRNA,但是在某些数据集中,rRNA最多可以代表80%的测序读段。
问题6:鉴定了多少rRNA序列?现在还剩下几读?
步骤6.复制
去除污染物,宿主序列和rRNA后,我们需要将以前去除的重复读段替换回我们的数据集中。
./3_Reduplicate.py mouse1_qual.fastq mouse1_unique_mRNA.fastq mouse1_unique.fastq.clstr mouse1_mRNA.fastq
注意事项:
该脚本的参数结构为: 3_Reduplicate.py <Duplicated_Reference_File> <Deduplicated_File> <CDHIT_Cluster_File> <Reduplicated_Output>
问题7:确定了多少假定的mRNA序列?有多少个独特的mRNA序列?
既然我们已经过滤了载体,衔接子,接头,引物,宿主序列和rRNA,请使用FastQC检查reads质量:
fastqc mouse1_mRNA.fastq -t 4
mouse1_mRNA_fastqc.html
问题8:滤出了多少总污染物,宿主和rRNA读数?
步骤7.分类
现在我们有了推定的mRNA转录本,我们可以开始推断我们的mRNA读数的来源了。首先,我们将尝试使用基于引用的短阅读分类器来推断我们阅读的分类起点。在这里,我们将使用[Kaiju](https://github.com/bioinformatics-centre/kaiju)基于参考数据库为我们的阅读生成分类学分类。Kaiju可以在内存少于16GB(约13GB)的系统上使用proGenomes数据库,以每分钟数百万次reads的速度对原核生物读物进行分类。使用整个NCBI nr数据库作为参考大约需要43GB。同样,快速分类工具需要大于100GB的RAM才能对大型数据库的reads进行分类。但是,Kaiju对于车间中的系统仍然占用了过多的内存,因此我们已经预先编译了分类,mouse1_classification.tsv,在tar文件中precomputed_files.tar.gz。
tar --wildcards -xzf precomputed_files.tar.gz kaiju*
chmod +x kaiju*
tar -xzf precomputed_files.tar.gz mouse1_classification.tsv nodes.dmp names.dmp
注意事项:
您将使用的kaiju命令如下:
./kaiju -t nodes.dmp -f kaiju_db.fmi -i mouse1_mRNA.fastq -z 4 -o mouse1_classification.tsv
命令行参数是:
-t:分类ID的层次表示
-f:kaiju的预先计算的索引
-i:输入内容为
-z:系统上支持的线程数
-o:kaiju分类标准的输出文件
然后,我们可以进行分类阅读并进行补充分析。首先,我们将分类的特异性限制在属级分类单元上,这限制了虚假分类的数量。
./4_Constrain_Classification.py genus mouse1_classification.tsv nodes.dmp names.dmp mouse1_classification_genus.tsv
注意事项:
该脚本的参数结构为: 4_Constrain_Classification.py <Minimum_Taxonomic_Rank> <kaiju_Classification> <nodes_file> <names_file> <Output_Classifications>
然后,我们使用Kaiju生成人类可读的分类摘要。
./kaijuReport -t nodes.dmp -n names.dmp -i mouse1_classification_genus.tsv -o mouse1_classification_Summary.txt -r genus
注意事项:
命令行参数是:
-t:分类ID的层次表示
-n:与每个分类ID对应的分类名称
-i:海归类分类
-o:摘要报告输出文件
-r:将为其生成摘要的分类等级
问题9:kaiju分类了几读?
最后,我们将使用[Krona](https://github.com/marbl/Krona/wiki)生成数据集分类组成的分层多层饼图摘要。
./kaiju2krona -t nodes.dmp -n names.dmp -i mouse1_classification_genus.tsv -o mouse1_classification_Krona.txt
tar -xzf precomputed_files.tar.gz KronaTools
sudo KronaTools/install.pl
KronaTools/scripts/ImportText.pl -o mouse1_classification.html mouse1_classification_Krona.txt
然后,我们可以使用网络浏览器查看此数据集的饼图表示形式:
mouse1_classification.html
看这张交互式的图还是很漂亮的。
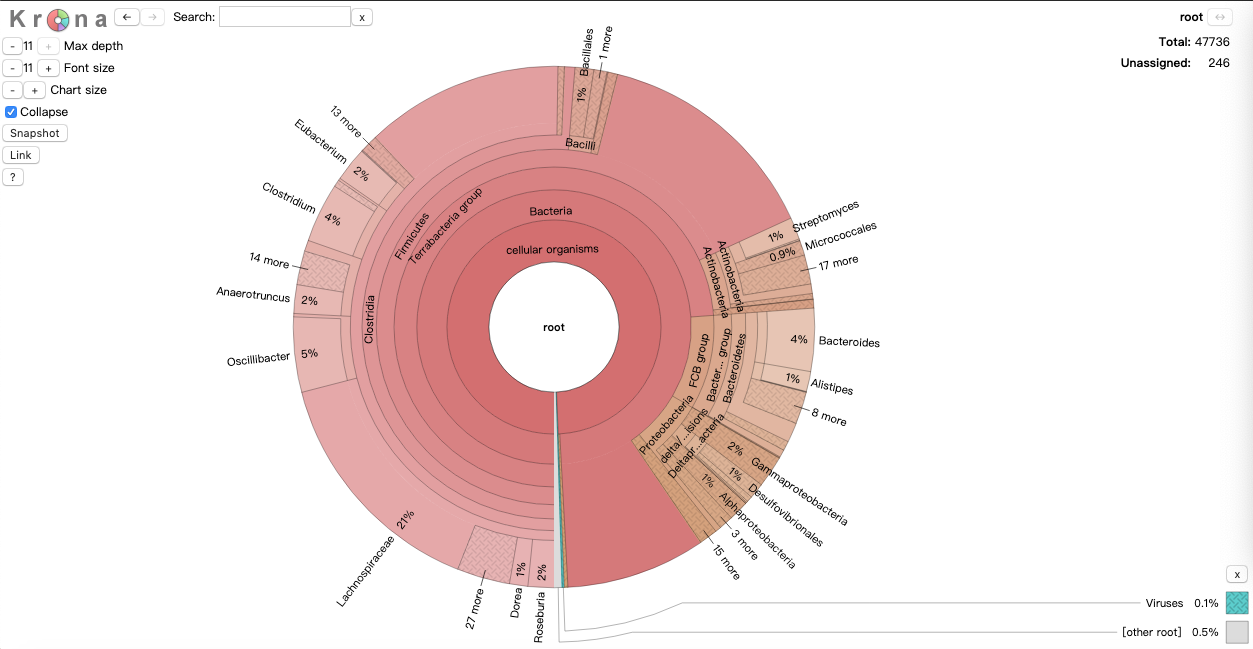
问题10:在我们的数据集中,最丰富的家庭是什么?什么是最丰富的门?
提示:尝试减小Max depth屏幕左上方的值,和/或双击特定的分类单元。