Literature-Based Genetic Risk Scores for Coronary Heart Disease
摘要
背景
全基因组关联研究(GWAS)已经鉴定出许多与冠心病(CHD)或冠心病危险因素(RF)相关的单核苷酸多态性(SNPs)。使用前瞻性心血管注册Maastricht(Carema)队列中的病例队列研究,我们测试基于GWAS鉴定的SNP的遗传风险评分(GRS)是否与未来CHD相关并预测未来CHD。
方法和结果
事件病例(n=742),即在中位数随访12.1年(范围0.0-16.9年)期间发展为CHD的参与者,与从总队列中随机选择的2221名参与者(n=21 148)进行比较。截至2011年5月2日,我们在GWAS中对先前与CHD或CHD RF相关的179个SNP进行了基因分型。由全部SNPs、153个RFSNPs或29个CHD SNPs组成的等位基因计数GRS与CHD无关。加权的29个CHD SNP GR,每个SNP的权重从GWAS获得,与冠心病无关(危险比,每个加权风险等位基因1.12;95%置信区间,1.04-1.21)和改进的风险重新分类2.8%(P=0.031)。作为一种实现加权的探索性方法,我们对所有SNP和CHD SNP进行了最小绝对收缩和selection operator(LASSO)回归分析。CHD套索GRS表现等同于加权CHD GRS,而整体套索GRS表现略好于加权CHD GRS。
结论
针对SNP的效应大小进行调整时,由CHD SNP组成的GRS改善了风险预测。或者,可以使用套索回归分析来实现加权;然而,需要在独立群体中进行验证。
简介
冠心病(coronary heart disease,CHD)是一种既受生活方式影响又受遗传因素影响的复杂疾病。在过去的几年中,全基因组关联研究(GWAS)已经确定了与CHD风险或CHD风险因素(包括血压和血脂水平)密切相关的多个共同单核苷酸多态性(SNPs)。希望GWAS将识别在预测CHD风险中有用的SNPs。然而,单独而言,这些SNP具有相对较小的效应大小。对于包含单个SNP累积效应的遗传风险分数(GRS),可能期望更大的效应大小。
之前的两项前瞻性研究调查了GRS与CHD 的相关性,发现基于已知与心血管疾病(CVD)相关的101个已发表的GWAS SNP及其中间风险因素的等位基因计数GRS与美国妇女的CVD相关,但与ATP III或Reynolds风险评分中使用的风险因素无关。基于12个SNP的子集选择与临床心血管终点相关的等位基因计数GRS的关联性稍强。在最近的芬兰队列中,基于13个GWAS鉴定的CHD相关SNP的加权GRS与冠心病的10年发病率独立相关。
在我们的研究中,我们基于截至2011年5月2日发表的GWAS中与CHD或CHD风险因素相关的179个SNP,组成了3个等位基因计数GRS;即基于所有179个SNP的总体GRS,基于与CHD风险因素相关的153个SNP的风险因子GRS,以及基于与CHD相关的29个SNP的等位基因计数CHD GRS。此外,用从2个已发表的GWAS获得的权重构建了一个加权CHD GRS。作为探索性分析,我们基于所有179个SNP和29个CHD SNP构建了最小绝对收缩和选择算子(LASSO)GRS。我们在以人群为基础的环境中调查了它们对冠心病的预测价值,平均随访时间为12.1年。
方法
SNP选择
使用GWAS Catalog16(www.genome.gov/gwastudies;2011年5月2日访问),在至少2个GWA研究中或在全基因组显著水平的荟萃分析(P<5*10−8)中,确定了与冠心病或其中间风险因素(即血压、人体特征(体重指数、腰围和腰臀比)、血脂水平和2型糖尿病)相关的SNP。对于彼此处于完全连锁不平衡(LD)(D‘=1;R2=1)的SNP,排除其中1个(n=22)。对于29个SNP,不可能设计引物或它们不适合分析设计,留下197个SNP用于基因分型(见在线数据补充表I)。
统计分析
为了估计缺失基因型(1.76%),使用了多重填充方法(R Packages mi v0.08-08)。缺失的基因型被归因于,其中成为主要纯合子、杂合子或次要纯合子的机会取决于特定SNPs的分布。这个过程重复了20次;因此,创建了20个具有不缺失数据的新数据集。接下来,按如下所述分析所有输入的数据集,并使用R package mitools(2.0版)组合来自每个输入数据集的结果。有关非推定数据集的结果,请参阅仅联机数据补充表IV。
我们构建了3个等位基因计数GRS和1个加权GRS。第一个GRS包括了与CHD相关的所有179个SNPs及其中间风险因素(总体GRS)。这种GRS背后的基本原理是将所有与CHD或其中间风险因素相关的已知SNPs的信息结合起来,并在前瞻性队列研究中测试其预测价值。第二组由153个SNPs组成,这些SNPs与CHD(风险因子GRS)的中间危险因素相关。包括这个GRS是为了评估这些SNP可以在多大程度上取代TRF(traditional risk factors)。第三个GRS由先前与CHD相关的SNPs组成(CHD GRS)。包括这个GRS的原因(没有被SNP稀释,对TRF的影响很小)是为了测试这个GRS是否会与CHD事件相关,并可能除了TRF之外还可能改善风险预测。Paynter等人使用了类似的方法,尽管当时发现的与CHD或其中间风险因素相关的SNPs较少。第四个GRS包含相同的29个CHD SNP,但每个SNP根据其效应大小(加权CHD GRS)进行加权,以允许预测主要基于最具信息性的SNP。为了估计单个SNP的权重,从2个大型GWAS中获得了对CHD的效应大小(见仅在线数据补充表III)。Ripatti等人使用了类似的方法。
我们测试了这4个GRS是否与未来的CHD相关,使用具有稳健方差估计的Cox比例风险模型根据Prentice方法对病例队列设计进行调整。这些模型用于估计CHD的10年风险,使用有和没有每个GRS的基础模型。基本模型由这些TRFs组成:性,当前吸烟,自我报告的糖尿病,父母的MI史,HDL胆固醇,总胆固醇,收缩压和BMI。年龄(以年为单位)被用作时间尺度变量,我们对延迟输入进行了调整。对每个SNP也分别进行相同的分析。
由于比例的变化(等位基因计数CHD GRS理论范围为0至58,而加权CHD GRS范围为0至22.14),加权GRS的风险比不能直接与等位基因计数GRS进行比较。因此,为了比较不同GR的效果,我们计算了所有个体中所有GR的z得分,并对这些标准化GR进行了上述分析。
为了测试向基本模型中添加GRS是否改善了风险预测,使用了c统计量,根据DeLong等人20的非参数方法和Pencina等人所描述的净重新分类改进(NRI)。NRI不直接适用于我们的研究,因为非病例(n=550)被审查(例如,在10年随访期内由于CHD以外的原因死亡)和病例(n=208)在10年随访后发生事件。因此,在10年随访后发生事件的病例被视为在10年随访时离开研究的对照对象(n=208),这导致434例和2012例无病例。接下来,我们计算子队列的Kaplan-Meier曲线。基于这个Kaplan-Meier曲线,权重被分配给每个个体。零权重分配给在随访10年前离开研究的非病例(n=550)。使用10年风险估计,将受试者分为以下风险类别:0%至<5%,5%至<10%,10%至<20%,和≥20%,如先前应用的。对于所有分析,我们根据Prentice和Kaplan-Meier曲线,使用软件R(版本2.10.1,www.r-project.org)结合软件包survival(版本2.35-8)用于Cox比例风险模型。为了估计c统计量和相应的概率值,我们使用了R package proc(版本1.4.1)。
基于LASSSO回归的GRS
在我们基于人群的前瞻性研究中,并非所有GWAS识别的SNPs都可能导致未来CHD的风险。23因此,作为一种探索性分析,我们使用套索回归选择了信息量最大的SNPs子集,24结合10倍交叉验证来解决内部验证问题。使用套索方法,从29个CHD SNPs(CHD Lasso GRS)和完整的SNPs集合中提取了信息性SNPs子集两个GR都接受与前四个GR相同的统计分析。对于套索回归分析,使用R Package penalized(版本0.9-32)。有关此分析的更多详细信息,请参阅仅限在线的数据补充方法。
结果
GRS
由所有的179SNP构建了总体GRS,其中的详细概述包括它们与TRF调整前后的CHD的关联,在仅在线的数据补充表IV中提供。所有参与者的总体GRS的平均值(SD)为182.9(9.2),其范围从142至217个风险等位基因。如表2所示,总体GRS与CHD相关,每个危险等位基因的危险比(HR)为1.02(95%置信区间[CI],1.01-1.03);调整TRF后,效应大小进一步减弱(HR/危险等位基因,1.01;95%CI,1.00-1.02)。
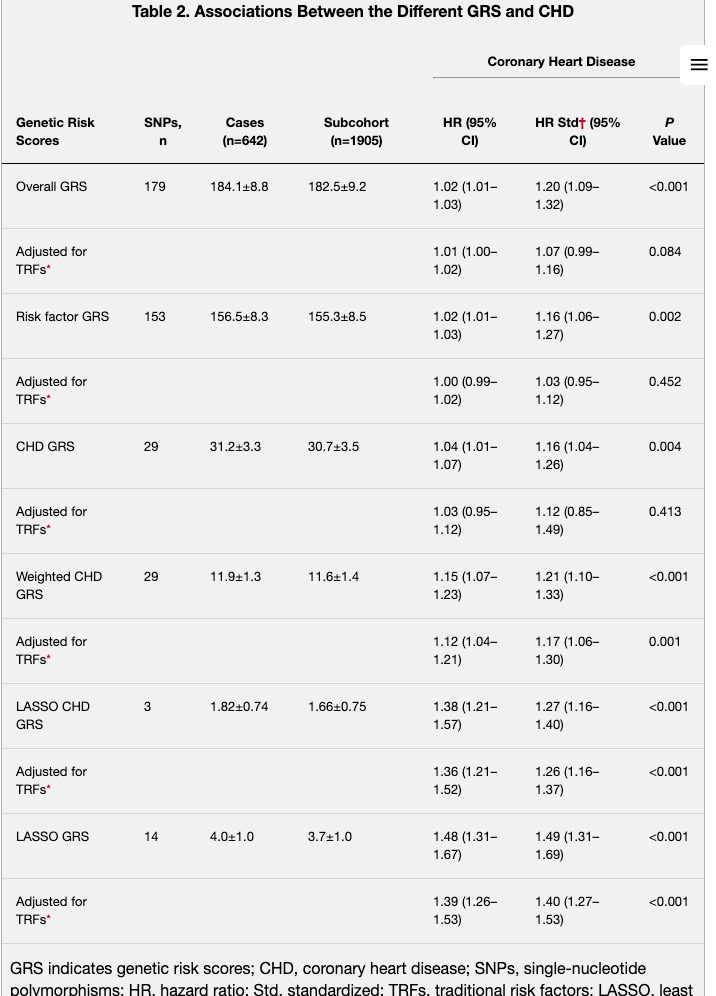
如表2所示,由153个SNP构建的风险因子GRS与未来CHD相关(HR/Risk等位基因,1.02;95%CI,1.01-1.03),但不是在调整TRF(HR/Risk等位基因,1.00;95%CI,0.99-1.02)之后。
接下来,调查从29个SNP构建的CHD GRS,这些SNP是为它们先前与CHD的关联而选择的。等位基因计数CHD GRS与未来CHD相关,每个危险等位基因的HR为1.04(95%CI,1.01-1.07)。调整TRF后,此GRS不再关联(HR,1.03;95%CI,0.95-1.12)。然而,在TRF调整之前(HR,1.12;95%CI,1.04-1.21),对先前报告的包含SNP的效应大小进行加权的CHD GRS与未来CHD相关(HR,1.15;95%CI,1.07-1.23)。
C-Indexes
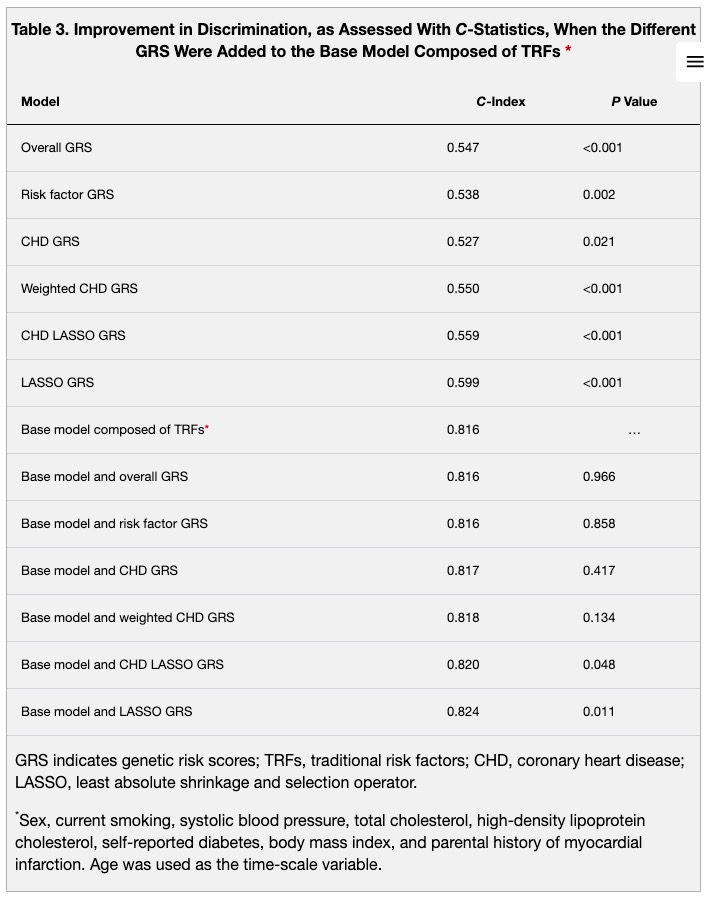
不包括其他变量的4个GRS的判别能力较低(总体GR的c指数=0.547,危险因素GR的c指数=0.538,冠心病GR的c指数=0.527,加权的CHD GR的判别能力为0.550),但都具有统计学意义(见图E和表3)。如图和表3所示,当使用基于所有TRF(c-指数=0.816)的预测模型时,添加4个GRS中的任何一个都不能改善风险辨别。

Net Reclassification Improvement
我们发现,当总体GRS、风险因素GRS或等位基因计数CHD GRS添加到基础模型中时,重新分类没有改善(表4,有关更多详细信息,请参阅仅限在线的数据补充表VII,A到C)。当我们使用加权冠心病GRS时,8.1%的参与者被归入更合适的风险类别,但对于5.3%的人口,重新分类变得更糟;因此,NRI提高了2.8%(P=0.031),这可以归因于事件的重新分类的改进(即,有冠心病事件的个体;NRI=2.5%,P=0.040,参见仅在线数据补充表7,D)。
基于LASSSO回归的GRS
作为探索性分析,我们对先前与CHD相关的29个SNP进行套索回归分析,提取出3个具有相应权重的SNP。这些SNP及其权重用于组成加权GRS(有关包含的SNP及其相应权重,请参阅仅在线数据补充结果和仅在线数据补充表VI)。这种CHD套索GRS与发生CHD相关(HR,1.38;95%CI,1.21-1.57),并且在调整TRF后仍然显著相关(HR,1.36;95%CI,1.21-1.52)。此CHD套索GRS的标准化HR(HR/SD增加,1.27;95%CI,1.16-1.40)与加权CHD GRS的标准化HR(HR/SD增加,1.21;95%CI,1.10-1.33)相当。接下来,我们对所有SNP进行套索回归分析,结果提取了14个具有相应权重的SNP(有关包含的SNP及其权重,请参阅仅在线数据补充结果和仅在线数据补充表VI)。基于这14个SNPs及其权重的套索总GRS与CHD发病相关(HR,1.48;95%CI,1.31-1.29),并在调整TRF后保持显著关联(HR,1.39;95%CI,1.26-1.53)。如表2所示,套索GRS的标准化HR高于加权CHD GRS的标准化HR。
部分讨论
从我们的研究中,我们可以得出以下结论。首先,由先前与CHD和/或中间CHD风险因素相关的共同SNP组成的3个等位基因计数GRS目前在预测冠心病发病方面没有价值,与在单个时间点测量经典生化参数和收缩压相比没有价值。第二,考虑到GRS中SNPs的不同效应大小可能很重要,考虑到在对TRF进行调整后加权CHD GRS仍然与CHD相关,并且等位基因计数CHD GRS不相关。这些结果与Davies等人的一项研究一致,23他们在研究中表明加权GRS优于等位基因计数。第三,当使用除TRF之外的加权CHD GRS时,当考虑到使用新的预测因子也会恶化重新分类这一事实时,2.8%的参与者被置于更合适的风险类别中。第四,10倍交叉验证套索回归可能用于从更大的SNPs集合中提取SNPs子集,尽管这种方法必须在独立的前瞻性队列研究中进行外部验证。
总之,与等位基因计数CHD GRS相比,加权CHD GRS与独立于TRF的未来CHD相关,并改善了风险重新分类。我们的结论是,重要的是调整不同SNP的效应大小。如果没有关于效应大小的信息,可以使用套索回归分析来估计这些效应大小。这种方法必须在独立前瞻性队列研究中进行外部验证。