Nature子刊:基于扩增子或宏基因组测序,预测菌群代谢组
Nature Communications[IF:11.878]
① MelonnPan是从菌群特征预测其代谢组的计算框架,给予模型上训练配对的代谢组和宏基因组,在新的菌群环境中预测潜在不可观察的代谢产物;② 将MelonnPan应用于两个独立的肠道宏基因组数据集,共220例受试者包括溃疡性结肠炎、克罗恩病和健康对照,在>50%的代谢产物中预测和观察到的菌群代谢趋势之间高度一致;③ 应用在珊瑚、鼠肠道和人阴道的微生物组的扩增子中,也保持相似的准确性;④ 还能提供预期性能评分,指导模型在新样本中的应用。
Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences
07-17, doi: 10.1038/s41467-019-10927-1
【主编评语】分析菌群代谢组有助于阐释菌群功能,但是对大规模样本进行代谢组学分析还是比较困难且昂贵的。Nature Communications近期发表一项研究,开发了一种算法,可通过机器学习,基于宏基因组或扩增子数据来预测菌群的代谢组,值得专业人士关注。(@李丹宜)

热心肠研究院的这个介绍让我对这个软件产生了好奇,我决定学习一下这个软件的使用,看看它和picrust的区别在哪,picrust2刚刚发布,看看是棋逢对手还是略胜一筹呢。后来发现,好吧,最后发现一个实验室开发的。。。区别在于一个是完全基于已知的参考数据库,而这个目标是发现是大多数(>60%)未注释基因家族与代谢物相对丰度的关联。
1.前言方面
介绍了代谢图谱预测的优点和其限制性,优点当然是节省成本啦,限制性在于:肠道微生物和代谢特征之间的关联强度表明,根据元基因组的分类或功能特征,可以大致预测微生物群落的代谢活性或特征。单纯基于酶的作用很容易识别这种关联,这在很大程度上受到目前基因-代谢物反应的不饱和组分以及典型测序和代谢组学技术提供的相对(而非绝对)丰度测量的限制。
最近的一些研究,在已知微生物群和代谢组学联系机制的情况下,对病例子集进行此类预测,例如基于化学计量酶反应矩阵的KEGG数据库。一组方法,统称为预测反应性代谢(Predicted Reactive Metabolic Turnover -PRMT)。另一组方法是基于约束或基于网络的建模框架,参考文献里好像没有picrust,大概都是一些建模的文章,基本上没有提供代码或者使用方法。
melonnpan通过以下方式推断复合代谢组:(1)数据驱动的最佳预测微生物特征集的识别;(2)对预测良好的代谢产物的预测准确度进行稳健量化。这使得研究人员可以重复地推断目前只有元基因组的群落的代谢物。
代码实现是一个R脚本,模型与实施方法文件在这:http://huttenhower.sph.harvard.edu/melonnpan
结果
1.算法
这是一种利用扩增子或宏基因组测序数据预测代谢物特征的计算方法,将生物学知识以分类或功能特征的形式结合起来。与现有的基于化学计量学的方法不同,这些方法依赖于数量有限、特征明确的分类群、酶和代谢物,功能注释对于melonnpan来说不是必要的,因为该工具的设计目的是通过机器学习(即使是从非特征化的微生物特征)来获取见解。在这篇文章里,我们讨论肠道微生物,但是方法学适应于任何微生物环境。简而言之,melonnpan使用弹性网络规则来确定哪些特征(分类或功能)是对给定代谢物的预测。给定一个新的分类图谱(来自扩增子或元基因组)或元基因组功能图谱(即基因家族Abun-Dances),然后结合序列特征的子集来估计相关的复合代谢组。所得预测代谢物均为预测特征(分类群或基因家族)相对丰度的加权和,其中训练弹性网络模型的回归系数用作预测算法的权重。
在优化阶段,既使用了测序数据,也使用了代谢组学实验数据。算法比较复杂,特别是对于计算机不够专业的人,于是这部分略过了。
2.在新群落中评估预测准确性
其比MIMOSA表现更出色,据我们所知,MIMOSA被评估为目前唯一能够根据人群水平的宏基因组数据预测全群落代谢相对丰度的方法,并提供了一个软件。MIMOSA建立在前面提到的代谢网络模型(PRMT)的基础上,从分类组成和宏基因组估算微生物群落的代谢潜力。简单地说,mimosa首先生成一个描述基因和代谢物之间定量关系的化学计量矩阵,以估计感兴趣群体的CMP评分。接下来,将所有样本对的CMP评分的差异与相应测得代谢物的差异进行比较。为了确定具有统计意义的预测良好的代谢物,MIMOSA依赖于基于Mantel检验的错误发现率(fdr)校正的P值来确定两个距离矩阵之间的相关性。类似地,它依赖用户提供的代谢物和微生物序列特征配对表。然而,与Memonpan不同的是,它没有明确地使用数据挖掘和模型构建来构造和验证预测模型。因此,MIMOSA无法对该算法之前未发现的新的元基因组样本进行预测。
Mimosa只能很好地预测少量代谢物化合物(补充图8a;n=20(23%),Mantel检验q<0.05)。相比之下,Memonpan能准确预测绝大多数代谢物(n=130(84%),斯皮尔曼R>0.3)。这表明,注释的微生物酶活性的比例相对较小,与使用机器学习新识别的酶活性之间可能存在较大差距。
3.揭示了有意义的生物学关系
使用HUMAnN2将菌分类分配给基因家族,进行了基因集富集分析(GSEA)来鉴定代谢模型最常选择的基因类别。Pseudoflavonifractor, Clostridium, Coprococcus, Anaerotruncus, Blautia, Collinsella, Ruminococcus, 和 Anaerostipes最显著。除了Collinsella来自放线菌门,其余均来自厚壁菌门。梭状芽孢杆菌和瘤胃球菌优先定植粘液层,从而提高了结肠上皮细胞对丁酸盐的生物利用度。这些细菌相对丰度的下降和一些疾病,如IBD相关。Clostridium cluster IV是短链脂肪酸(SCFAs)产生菌,SCFAs被认为是肠道菌与宿主间的信号分子。
使用Pfam数据库,将这些代谢预测基因家族分类为蛋白质,令人惊讶的是没有个体pfam家族富集,在预测基因家族中有一个显著的未识别的蛋白域的过度表达。这些未识别蛋白可能在群落中具有潜在重要作用。
4.预测的代谢物揭示了IBD代谢组的整体结构
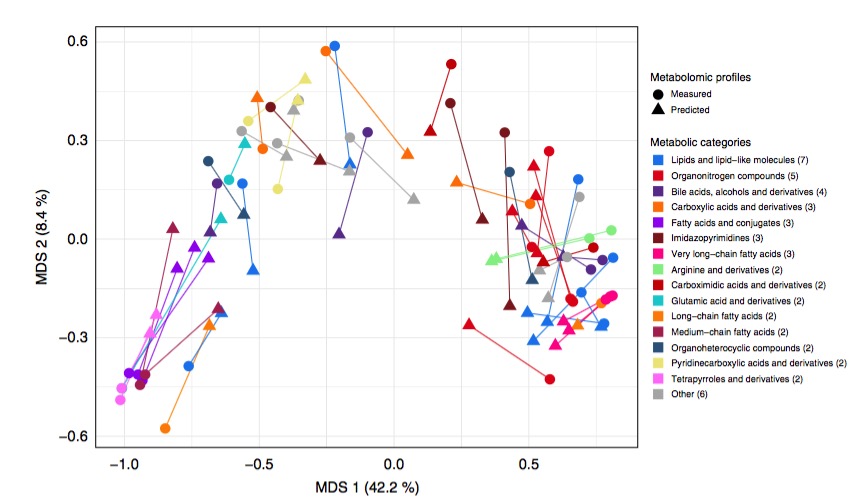
最近的几项研究表明,IBD患者和健康个体以及IBD亚型(UC和CD)可以通过代谢谱来区分。使用melonnpan发现的,与IBD相关的预测良好的代谢产物跨越了广泛的代谢类别,包括氨基酸、胆汁酸、脂肪酸和鞘脂等。大约三个聚类显著:1)在IBD中耗尽的几种胆汁酸和极长链脂肪酸基团;2)在IBD中富集的几种胆固醇和四吡咯衍生物;3)少量的非差异丰富的代谢产物,如氨基酸、肽、嘌呤及其衍生产物。基于测量和预测剖面的影响大小估计值之间的斯皮尔曼相关性:CD与HC分别为0.70,UC与HC比较分别为0.45;p<2.2e−06,提示即使在缺乏全面的代谢谱分析的情况下,melonnpan也可以从宏基因组推断与疾病相关的代谢组分差异。
与以前的研究一致,melonnpan预测IBD患者的初级胆汁酸显著升高,次级胆汁酸显著降低。胆汁酸的生物合成是由微生物酶活性直接介导的,在IBD中,微生物酶活性不能去除原代胆汁酸的结合物,导致次级胆汁酸及其对肠上皮细胞的抗炎作用的减少。与对照组相比,IBD受试者体内四吡咯类化合物的含量往往持续下降。综上所述,这些发现证实了Melonnpan能够在广泛的化合物中提供代谢相关的预测,识别肠道微生物群-代谢组学轴的重要贡献者,进而促进对微生物群的大规模综合多组学分析。
5.对人体和环境微观生物的推断。
最后,我们将Melonnpan应用于三对16s和代谢组学数据集,以说明Melonnpan能够从各种微生物环境和分析类型中获得生物学发现。样本分别来自珊瑚微生物群落,阴道微生物群落和小鼠肠道微生物样本。代谢组分别采用核磁共振,靶向LC-MS和非靶向LC-MS+气体色谱(GC-MS)。由于样本量较小,在每个数据集中分析的代谢物中,>50%未通过单独的预过滤,被舍弃。我们发现,在每一组数据中,超过60%的分析代谢物预测良好。这表明,对于大部分化合物而言,包含在一些分类特征中的信息足以解释代谢物丰度的大部分变化(和前面肠道基因家族应用一致)。值得注意的是,与人类肠道样本不同,我们无法在这些环境中访问独立的验证数据集,因此,这只是个初步评估。
讨论
尽管Melonnpan的预测方法不能取代代谢组学分析,但它可以以远小于代谢组学的成本来大致预测和比较许多样本中可能的代谢,从而使用现有的宏基因组数据研究提出假设和预测代谢组学。
为了指导用户使用,MelonnPan专门为每个新的微生物群落提供置信分数(RTSI),低置信分数表示与训练宏基因组的高度不同性。由于宏基因组之间的训练差异(由RTSI捕获)会影响MelonnPan的准确性,因此RTSI值可以用作指示在新环境中可能需要多少额外的代谢体数据来补充预先训练的MelonnPan模型。
由于与次生代谢物产生相关的主要微生物表型差异往往是物种或菌株特有的。具体地说,我们的分析证实了之前报道的几个IBD相关物种是肠道中微生物代谢物动力学的重要驱动因素。虽然物种水平的预测因子在训练队列中导致了类似的表现,但这些基于分类学的预测并没有遗传到独立队列。验证队列中的这种显著较低的可预测性可能反映了基因家族数据捕捉到的菌株水平效应,因为不同群体中的菌株差异可以显著影响代谢物预测的概括性。这突出了将基因水平谱作为预测因子的重要性,因为仅从物种丰度数据中可能无法捕捉到特定的菌株特异性代谢以及其他表型相关性状(例如抗生素抗性)。
这个方法的限制必须考虑到。MelonnPan不直接预测代谢物通量或峰值(与基于约束的方法相反);相反,它通过合成和组合微生物序列特征,提供对每个变质岩在整个群落范围内的相对丰度的估计。对于跨环境预测来说,没有一个模型是准确的。因此,将其作为一种假设生成工具,鉴于一般一致性通常足以为后续的实验验证研究提供依据,但这些研究必须实验确认完全符合预测并获得直接数据。
有趣的是,即使是独立于机制和分子起源做出的预测,某些特定代谢物的强可预测性可能在暗示这些机制方面也有价值。因此,从培养无关的群体水平数据进行鉴定的未来工作有可能侧重于菌株特异性基因集,甚至可能是生物活性分类群之间的单核苷酸多态性水平差异。进一步细化MelonnPan预测精度的未来研究的其他方向包括:(I)整合其他类型的微生物测量,例如宏转录数据,(Ii)利用纵向剖面的动态预测,(采用更复杂的机器学习策略,如多变量或贝叶斯框架,这些策略可以明确纳入定量特征,如全社区酶特异性反应信息和零膨胀等。
宏基因组学测序已经研究了包含数百万个分类群的数万个样本和微生物基因,基中的数以百万计是没有被表征的。例如,UniProtKB中仅有约1.0%的所有蛋白质已被实验表征。因此,MelonnPan的人类肠道模型的一个重要发现是大多数(>60%)未注释基因家族与代谢物相对丰度的关联。基因家族和代谢物之间的这种联系为基因本身的下游特征描述提供了有前景的目标,特别是当应用于其他特征不太明显的环境时,因为它们可能在这些化合物的生成或代谢中起作用。因此,这种计算方法(i)生成生化和功能基因组假说,以供将来验证,(ii)有助于系统范围内对微生物群的理解。(iii)为现有代谢重建模型提供了一个补充工具,(四)有助于为代谢组学在微生物群落中的转化应用,并奠定实验设计基础。随着参考数据库的继续更新,以及训练数据集的扩展,MelonnPan的预测精度将默认随着时间的推移而提高。综上所述,这一分析框架是迈向群体水平宏组学数据整合的必要的第一步,最终使我们能够更好地理解微生物组的动态,超越分子目录,进入微生物组研究的健康应用。